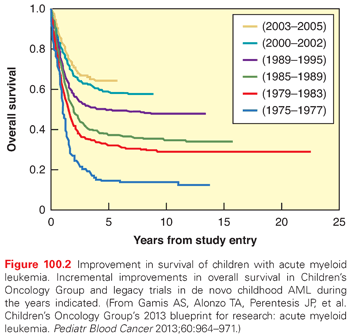
Acute myeloid leukemia (AML) accounts for approximately 15% to 20% of childhood leukemias, chronic myelogenous leukemia (CML) accounts for 3% to 4%, and juvenile myelomonocytic leukemia (JMML) and other rarer histologies account for less than 1%. The incidence of AML is currently estimated to be 5 to 7 cases per million in the United States.15 Myelodysplastic syndrome (MDS) is a relatively rare diagnosis in childhood, generally progresses to AML, and is treated with similar regimens (including bone marrow transplant) as AML.16 AML incidence is slightly higher in African Americans and Hispanics in the United States, and survival is slightly lower.17,18 Because of intrinsic differences in drug sensitivity between ALL and AML cells, progress in treating AML has been less dramatic than that of ALL. Nevertheless, current cure rates in AML have risen to approximately 45% to 65% with the advent of more intensive conventional chemotherapy and bone marrow transplant (BMT) (Fig. 100.2).19,20
Etiology
Environmental Factors
There is extensive literature and a continued interest in exploring possible relationships between infectious or environmental exposures and increased risk of childhood leukemia. However, most studies show weak or no correlation of these factors with the incidence of leukemia.21–23 Electromagnetic fields have not been found to be significant in pediatric malignancies.24–26 There is an emerging body of evidence from population-based studies of polymorphisms of drug-metabolizing genes, such as NQO1 and GST polymorphisms, that may be associated with an increase or decrease in an individual’s risk for developing leukemia based on exposures to particular environmental toxins like benzene and other organic solvents, quinine-containing substances, and flavonoids.27
Genetics
There is significant evidence that genetic factors play a role in the etiology of pediatric leukemia. Within the leukemic blasts themselves, there are characteristic cytogenetic changes (Table 100.1), many of which have prognostic significance (see later discussion). Key genetic changes found in childhood leukemia (especially in younger patients) may occur in utero or very early in life.28,29 Evidence includes the detection of cells bearing these changes in neonatal blood spots30 and transmission of leukemia by twin–twin transfusion.31 The delayed infection hypothesis holds that children in developed countries are spared the frequent early infections that are necessary for normal development of the immune system. An infection occurring at a later time is then hypothesized to elicit a pathologic, myelosuppressive response, providing a selective growth advantage to the preleukemic clone, leading to development of overt leukemia.32

In most cases, leukemia appears to result from a complex interplay between environmental and genetic factors. Recently, genome-wide association studies have identified several constitutional genetic variations with moderate but statistically significant effects on the relative risk of ALL.33–36 Collectively, variants in IKZF1, ARID5B, CEBPE, and CDKN2A may account for up to 80% of the attributable risk of developing ALL in Europeans.35 ARID5B and GATA3 polymorphisms have been identified as associated with both ALL susceptibility and relapse hazard, and may contribute to racial disparities in outcome, because high-risk alleles occurred more frequently in subjects with a greater degree of Native American ancestry.37
In addition to somatic and germ-line genetic changes associated with leukemia, there are well-described cases of familial leukemia, including recently described mutations in the hematopoietic transcription factor PAX5,38 as well as strong associations between leukemia risk and immunodeficiency and several somatic genetic disorders (e.g., Down syndrome [DS], Bloom syndrome, ataxia telangiectasia, Shwachman-Diamond syndrome, Noonan syndrome, neurofibromatosis).8 Recently, a new association has been described between Li-Fraumeni syndrome, a well-known cancer predisposition syndrome, and hypodiploid ALL.39
Patients with DS have a 10- to 20-fold increased risk of developing leukemia.40 The occurrence of leukemia in DS patients appears to be unrelated to the other congenital abnormalities and medical problems of DS.41 The cytogenetic changes common in ALL occur less frequently in DS-ALL,42–45 but two alterations are markedly enriched: (1) Janus kinase 2 (JAK2) activating mutations in approximately 20% of DS-ALL, and (2) rearrangements leading to overexpression of cytokine receptor-like factor 2 (CRLF2) in up to 50% of DS-ALL.46–48
In the neonatal period, DS patients have a propensity for developing a nonneoplastic entity known as transient myeloproliferative disease (TMD) of DS, which at presentation, can appear very similar to AML (i.e., very high white blood cell counts with peripheral myeloblasts), but usually resolves spontaneously.49 TMD may be difficult to distinguish from AML. Although TMD itself is not a form of malignancy or leukemia, approximately 30% of the DS patients with TMD will develop AML later in childhood.49
When patients with DS do develop AML (especially those younger than age 2 years), they tend to develop acute megakaryoblastic leukemia (AMKL), display particular mutations in the GATA-1 hematopoietic transcription factor, and have an excellent prognosis compared to other adult and pediatric AML subgroups. The disease-free survival (DFS) for DS patients with GATA-1+ AMKL is over 90%, with regimens significantly less intensive than those required for other AML cases.50 The reasons behind the increased risk of leukemia in DS remain unknown.
Clonal Nature of Lymphoid Cancers
Lymphoid malignancies appear to have derived from an original abnormal progenitor that lost the ability to fully differentiate and formed a clone of leukemic blast cells. ALL blasts are characterized by early cytoplasmic and surface lymphoid antigens (detectable by flow cytometry), as well as incomplete immunoglobulin (Ig) and T-cell receptor (TCR) gene rearrangements, suggesting they arise from a precursor T- or B-lymphoid progenitor. AML may arise from a multipotent or committed myeloid progenitor. Persistence of subclinical amounts of leukemia during or after therapy is called minimal residual disease (MRD).51 MRD may be detected by flow cytometry or polymerase chain reaction (PCR)-based detection of Ig/TCR rearrangements or chromosomal translocations.52 Use of high-throughput sequencing technologies for MRD detection is being undertaken on a research basis and may hold promise as a future clinical test with even higher sensitivity and precision.53
Diagnosis
The presenting signs and symptoms of a child with leukemia reflect the impact of bone marrow infiltration, the extent of extramedullary disease spread, and problems arising from changes in blood viscosity and chemistry related to the size, number, and breakdown products of leukemic blasts (tumor lysis).12 Leukostasis from high white blood cell (WBC) counts may cause signs and symptoms ranging from mild respiratory symptoms and pulmonary infiltrates, to stroke, cranial or peripheral neuropathies, hematuria, renal failure, and ocular findings.8 AML blasts are larger, less flexible, and contain granules that can cause inflammation and clotting. Thus, AML has a higher risk of serious systemic problems then ALL for a given high WBC (e.g., >100,000/mm3). Gingival infiltration and orbital and periorbital soft tissue masses (chloromas) are often seen in AML.54 AML FAB subtypes 4 and 5 have a tendency to present with chloromas.
Central nervous system (CNS) involvement is more common in ALL but is also possible with AML.55,56 CNS involvement is most often meningeal involvement, but may manifest as cranial nerve involvement, and occasionally, frank leukemic infiltrates in the parenchyma of the brain. CNS involvement may be asymptomatic or manifest as cranial nerve palsies, seizures, or focal neurologic findings. Testicular involvement generally manifests as a painless, palpable mass. Hepatosplenomegaly is common in both ALL and AML, and may compromise respiration in small children. Lymphomatous involvement of nodes or extranodal tissue can cause superior mediastinal or superior vena cava syndromes, and bowel wall or mesenteric node infiltrations can cause intussusception or bowel perforation.
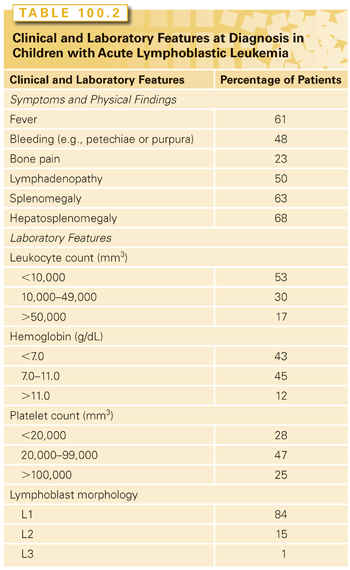
Tables 100.2 and 100.3 summarize the clinical and laboratory findings and differential diagnosis, respectively, for pediatric patients presenting with ALL. Infection and bleeding are significant risks. Fever should be managed aggressively with intravenous broad-spectrum antibiotics. Bleeding risk should be addressed by prompt transfusions of packed red blood cells, platelets, and/or plasma, as clinically indicated.

A definitive diagnosis of leukemia requires that the bone marrow has more than 20% to 25% leukemic blasts (depending on the pathologic classification system used).57 Traditionally, the diagnosis depended on bone marrow aspirate and biopsy morphology and immunohistochemical staining patterns. The cornerstone of modern diagnosis is immunophenotyping, using flow cytometry antibody panels (see Chapter 107) to define the lineage of the leukemic clone. Cytogenetics also play a critical role.
Other diagnostic approaches that have become standard in many centers include fluorescent in situ hybridization (FISH), and PCR (to identify specific fusion proteins arising from chromosomal rearrangements). Finally, several techniques are used primarily on a research basis, such as RNA expression arrays, DNA copy number and single-nucleotide polymorphism (SNP) arrays, and whole-genome and whole-exome sequencing.58–61 These techniques will be used increasingly for clinical decision making as well, such as to identify cryptic, actionable mutations62 or to predict susceptibility to targeted therapy.63,64
Management
Prognostic Factors in Management of Pediatric Acute Lymphoblastic Leukemia
As outcomes with modern therapy in pediatric ALL have improved, many factors previously shown to be important for predicting a prognosis have lost statistical significance. Five factors have retained prognostic significance and constitute the basis on which patients are stratified in most treatment protocols. These factors are (1) age at presentation, (2) WBC at presentation, (3) specific cytogenetic abnormalities, (4) presence or absence of CNS involvement (Table 100.4), and (5) rapidity of initial response to chemotherapy.8 The 1996 National Cancer Institute (NCI) consensus criteria standardized definitions of age, initial WBC, and CNS involvement to facilitate a comparison of clinical trial results between groups.65 Cooperative groups differ in incorporation of cytogenetics and response to therapy. General principles of modern ALL protocols include treatment of low-risk disease with less-intensive chemotherapy to minimize toxicity while maintaining excellent overall survival (OS) (90% to 95%), whereas high-risk disease (OS, 40% to 85%) is treated with more intensive therapy.8

The age-determined risk groups are less than 1 year (infant ALL), 1.0 to 9.99 years (standard risk ALL), and ≥10 years (high-risk ALL). Infants constitute a very high-risk group. MLL (11q23) rearrangements are frequent, especially t(4;11)(q21;q23), and are associated with hyperleukocytosis, extramedullary disease, and common acute lymphoblastic leukemia antigen (CALLA) (CD10) negativity.66,67 Cure rates on current infant protocols have improved modestly to approximately 50%.68,69 Results are poorest in infants under 3 months of age with MLL rearrangement. Unlike for some other high-risk ALL subgroups, current evidence does not suggest a benefit of stem cell transplant.69
Adolescents and young adults have also traditionally demonstrated lower cure rates, although survival has improved significantly with early intensive postinduction therapy.12,70 In B-lineage ALL, presenting with a WBC count more than 50,000/mcL, and particularly over 100,000/mcL, is associated with a high risk of relapse.
Blast cell cytogenetics and ploidy have a significant prognostic impact in ALL (Fig. 100.3). High hyperdiploidy (≥50 chromosomes or DNA index >1.16) is associated with good prognosis, particularly with trisomies of chromosomes 4 and 10.71–73 Hypodiploidy (<45 chromosomes) and especially haploidy (23 chromosomes) are associated with a poor prognosis.72,74 Two other recently identified unfavorable abnormalities are IKZF1 alterations75 and intrachromosomal amplification of chromosome 21 (iAMP21).76 CRLF2 overexpression has been found to have an independent adverse prognostic impact in some studies, but not others.77,78
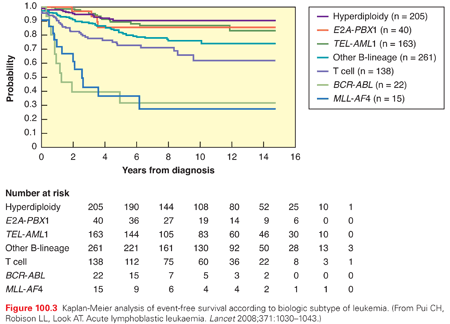
The most common translocation in pediatric ALL is t(12;21)(p12;q22), forming the ETV6-RUNX1 (also known as TEL-AML1) fusion. This cryptic translocation occurs in 25% of US pediatric ALL cases, less frequently in other geographic and ethnically defined populations, and is associated with a favorable prognosis.79,80 The Philadelphia chromosome (Ph+) refers to the t(9;22)(9q34;q11) translocation, forming the BCR-ABL1 fusion. Ph+ ALL is less prevalent in children than adults and was historically associated with a dismal prognosis. Treatment of Ph+ ALL and CML were revolutionized by imatinib mesylate, a selective tyrosine kinase inhibitor active against the BCR-ABL1 fusion. The addition of imatinib to conventional chemotherapy has improved survival from approximately 30% to 40% to 80%, and hematopoietic stem cell transplant (HSCT) in first remission is no longer considered as the standard of care.81–83 Ongoing studies are investigating the efficacy of later generation tyrosine kinase inhibitors such as dasatinib.84
Numerous translocations occur in T-lineage ALL, but they are not generally associated with a prognosis. Activating NOTCH1 mutations occur in over 50% of T-ALL cases, and are associated with a favorable prognosis.85,86 Because mutated NOTCH1 activity depends on γ-secretase activity, γ-secretase inhibitors are being investigated as a novel therapeutic approach. Early T-cell precursor (ETP) is a very unfavorable prognostic subgroup identified based on a distinctive immunophenotype, constituting approximately 10% of T-lineage ALL, which bears a gene expression signature and mutational profile similar to myeloid leukemia.61,87
CNS and testicular involvement are unfavorable prognostic signs, for which therapy is intensified.56 The CNS is considered a sanctuary site because many systemic treatments do not adequately penetrate the blood–brain barrier, so CNS preventive therapy is required in all patients to prevent eventual CNS relapse. CNS prophylaxis is usually achieved through systemic and intrathecal chemotherapy. Although CNS involvement at diagnosis has generally been treated in the past with the addition of radiation to intensive chemotherapy, some advocate for reserving CNS radiation for use only in the case of CNS relapse.88 Both testicular and CNS relapses require site-directed radiation as well as systemic reinduction due to increased risk of subsequent bone marrow relapse.12,89
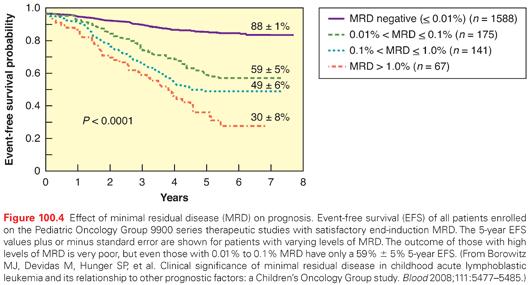
All modern pediatric ALL treatment protocols use the prognostic factors outlined here, in varying ways, to stratify patients into different risk groups that receive treatment of different intensity. The first phase of treatment (induction) lasts 4 to 6 weeks and includes a glucocorticoid, vincristine (VCR), asparaginase, and for high-risk patients, an anthracycline (usually daunomycin).12 The Berlin-Frankfurt-Munster group begins induction with a steroid window, and the degree of cytoreduction during this window is used in risk assessments.90 Some groups intensify induction with additional medications. Rapid response to induction is an important prognostic variable (Fig. 100.4).8,73 Induction failure (≥25% marrow blasts at the end of induction) is rare (2% to 5% of cases) and generally associated with a poor prognosis, although a recent large retrospective analysis reported an unexpected heterogeneity in survival, ranging from 10% to 70%, depending on age, cytogenetics, and B- versus T-lineage disease.91
Induction is usually followed by a consolidation phase to reinforce the bone marrow remission and to administer CNS prophylaxis. Subsequent intensive phases may be termed intensification, delayed intensification, or reinduction/reconsolidation, with the intensity depending on the risk group.12,92,93 These intensive courses are generally delivered within the first 6 to 12 months of treatment, followed by a maintenance or continuation phase lasting 2 to 3 years, which consists of antimetabolite treatment (usually consisting of methotrexate weekly and 6-mercaptopurine daily), periodic intrathecal (IT) treatments, and periodic pulses of VCR and oral glucocorticoids with the frequency of these pulses varying by protocol and treatment group. To minimize and avoid treatment-related toxicities, strict supportive care guidelines are employed. Therapy-related mortality on frontline pediatric ALL protocols is generally under 2% to 3%. Overall treatment duration is usually between 2 and 3.5 years. On current Children’s Oncology Group regimens, boys receive an additional year of maintenance, but many other cooperative study groups treat both genders for approximately 2 years.
CNS-directed therapy is usually present in all phases, but is most intensive during the first several months. CNS therapy uses intrathecal chemotherapy, high-dose systemic chemotherapy (principally, higher dose methotrexate or cytarabine), and/or cranial radiation.56,94 Because it is often associated with neurocognitive deficits, endocrine, and growth abnormalities, cranial radiation has generally been reserved only for patients at highest risk of CNS relapse (i.e., approximately 5% of patients who present with initial CNS involvement or T-cell disease), but at least one treatment group currently reserves the use of cranial irradiation only for CNS-relapsed cases.88 Through the years, the doses of cranial radiation have decreased from the 24- to 36-Gy range that was used on early protocols to 18 Gy for treatment and 12 Gy on some CNS preventive therapy regimens.56
Therapy for relapsed ALL depends on the location of relapse (isolated extramedullary, bone marrow, or combined) and the duration of initial remission. Extramedullary relapse generally has a better prognosis than bone marrow relapse, but requires localized in addition to systemic chemotherapy.12 For testicular relapse, local therapy involves orchiectomy, followed by irradiation of the remaining testicle and scrotal area.12,95 Fertility will be compromised (so postpubertal males should be offered sperm banking), and hormonal supplements should be offered as needed. Prognosis after a testicular relapse remains quite good. For CNS relapse, local therapy involves intensified intrathecal therapy, varying regimens of high-dose systemic therapy, and the addition of cranial and/or craniospinal radiation.
All relapse types have a worse prognosis if they occur during therapy or within 6 months of completing therapy.96 Interestingly, a recent study indicated that postrelapse survival does not differ according to the intensity of chemotherapy received prior to relapse.97 Multiple medications and regimens are being tested for induction in relapsed and refractory leukemia. A common approach is to administer several intensive blocks of conventional chemotherapy agents, with or without a concomitant novel investigational agent.98 Promising results have been recently obtained with clofarabine and clofarabine-containing regimens.99–101 Blinatumomab, a CD19/CD3-bispecific T-cell engaging (BiTE) antibody, has shown promising response rates and prolonged survival in relapsed B-lineage ALL in adults, and is currently in a phase II international trial in pediatrics.102,103 Promising recent results have been obtained using chimeric antigen receptor-modified T cells with specificity for CD19, termed CTL019 cells.104 The most recent update on this trial reported that among 20 patients (including 16 pediatric) with treatment refractory CD19+ ALL, 14 patients (82%) achieved complete remission, with robust in vivo expansion of CTL019 cells, and 11 of 17 evaluable patients maintained an ongoing CR during median follow-up of 2.6 months.105
The decision between chemotherapy and HSCT for second complete remission has been controversial. HSCT is usually used for relapse within 6 months of the completion of initial treatment (CR1 <30 months), but the decision also depends on suitable donor availability, the difficulty (number and intensity of induction attempts needed to achieve CR2), and the overall health of the recipient.106 Prior to HSCT, patients with relapsed ALL typically receive induction and consolidative chemotherapy, because the chances of success with HSCT are extremely low in the setting of continued active disease. Autologous BMT in ALL is no longer recommended.
Prognostic Factors and Management of Pediatric Acute Myeloid Leukemia
In contrast with ALL, there are fewer standard clinical or laboratory-based factors in AML that consistently relate to prognosis. Table 100.1 reviews prognostic factors in pediatric ALL and AML. Poor prognostic factors that predict lower remission rates and/or decreased event-free survival (EFS) include blast cytogenetics with monosomy 5 or 7, 5q deletion, FLT3/ITD, and secondary AML/MDS.19,107–109 Adolescents and young adults have an increased risk of treatment-related mortality, but no significant difference in OS.110 A swift response to induction chemotherapy (i.e., remission after one cycle of chemotherapy), favorable cytogenetics, and DS (with FAB M7) are predictive of better outcomes.108 Favorable cytogenetics in AML include core binding factor alterations (the t[8;21] AML1/ETO fusion and inv[16]), and the t(15;17) PML/RARα fusion seen in acute promyelocytic leukemia (APL) (see Chapter 107). Two other recently identified favorable cytogenetic alterations (CCAAT) are mutations in NPM1111 and CCAAT/enhancer-binding protein alpha (CEBP).112 In addition to genetic alterations, MRD is being integrated into current AML studies as an important prognostic marker.113 Although many findings in AML are similar between adults and children, important biologic and therapeutic differences are emerging. An example of this is that mutations of the KIT receptor tyrosine kinase have a similar frequency in pediatric core binding factor AML as those found in adult AML, but lack the poor prognosis seen in adults.109
There have been incremental improvements in AML outcomes over recent decades, with current overall survival in the 60% to 70% range (see Fig. 100.2). These improvements have come through increased intensity of therapy and improved supportive care.114,115 The inherent drug resistance of AML cells poses challenges because the intensification of therapy required is associated with significant treatment-related toxicity, particularly infections.19
With the exception of DS patients with FAB M7 AML and patients with APL, most pediatric AML induction therapies include two cycles of ara-C and daunomycin, with or without thioguanine and/or etoposide. Studies have shown that the intensity of induction therapy is important for OS.19 In the Children’s Cancer Group 2891 protocol, intensively timed induction (starting the second cycle on day 14 regardless of count recovery) did not change the induction (CR1) rate, but it did have a profound effect on eventual cure rate.116 Although fewer than 5% of pediatric AML patients present with CNS disease, as many as 20% will suffer an isolated CNS relapse.55 Intrathecal chemotherapy (usually ara-C) has been found by several groups to effectively reduce the risk of CNS relapse.19
Conventional postinduction consolidative chemotherapy in pediatric AML usually involves two to three cycles of high-dose ara-C–based combinations, to which anthracycline, etoposide, or ifosfamide/cyclophosphamide may be added. There is no proven benefit to maintenance chemotherapy, so conventional treatment is rarely more than four to eight cycles.19,117
Gemtuzumab ozogamicin (GO), a newer agent consisting of an antibody to CD33 coupled to calicheamicin (a toxic antitumor antibiotic), has shown some promise in relapsed AML and was recently tested in a randomized, prospective trial for frontline disease by the Children’s Oncology Group.118 GO was significantly associated with improved EFS, but not OS. Other adult and pediatric trials of GO in AML have yielded mixed results, and at present, it has been voluntarily withdrawn from the commercial market and its role in AML therapy remains to be determined. Two promising novel biologic agents, the proteasome inhibitor bortezomib119 and the kinase inhibitor sorafenib (which inhibits FLT3)120 are currently being tested for efficacy in combination with frontline conventional chemotherapy. The indications for HSCT in pediatric AML, preparative regimens, timing (CR1 or later), and type of donor are the focus of intensive ongoing controversy and research.116,121,122
Pediatric APL has among the highest cure rates in pediatric AML. These patients should receive all-trans-retinoic acid (ATRA), which directly binds to the t(15;17) translocation, which forms a fusion protein combining the promyelocytic leukemia (PML) gene with the retinoic acid receptor alpha (RARα) gene, which is causative for this form of AML.123 They should also receive conventional chemotherapy during both induction and consolidation phases. Patients who respond to this therapy have an 80% to 85% survival rate and should not be subjected to HSCT.124 Similar to adult therapies for APL (see Chapter 107), arsenic trioxide, found to be useful for salvage in patients who became resistant to ATRA, is currently being tested in upfront pediatric APL regimens (both alone and in combination with ATRA and conventional chemotherapy) in an effort to improve DFS in CR1.125 Rapid initiation of therapy is particularly important in APL due to the risk of severe bleeding complications in untreated disease. Early deaths from bleeding are often not captured on clinical trials, but population-based studies indicate that they are a significant cause of mortality.126 Despite the remarkable successes with ATRA and arsenic trioxide with and without conventional chemotherapy, there remain patients with APL with relapsed and refractory disease, and many of these can still be cured with HSCT.127
DS patients with AMKL (see earlier discussion) do well with lower dose ara-C regimens and standard timing of their induction cycles. Current results in DS AMKL are the best of any AML subgroup outside of APL, with 70% to 85% EFS, with patients under 2 years showing the best results, and thus not requiring intensive chemotherapy or HSCT.128–130
The prognosis for pediatric patients who relapse after either conventional or HSCT therapy for AML is poor. A second CR can be obtained using similar ara-C and anthracycline-containing regimens in 20% to 70% of patients, but the likelihood of obtaining a cure is approximately half the rate for de novo AML.131 Clofarabine alone and in combination (see ALL relapse discussion) with other chemotherapeutic agents also shows some efficacy.100,101 HSCT in early relapse or CR2 for AML patients who were previously transplanted in CR1 is a strategy that has had some success.131–133 Other novel therapies, including FLT3 inhibitors and hypomethylating agents, are currently being studied.134
Rarer Forms of Leukemia in Children
Chronic leukemias, with the exception of CML, do not occur in children. Ph+ CML is rare in childhood (<1% to 2% of all pediatric leukemia cases).135 When it does occur, it typically presents in adolescents and is in the chronic phase. Therapy is similar to that recommended in adults, with imatinib (along with hydroxyurea if the initial counts are high, and/or the patient presents with a high degree of hepatosplenomegaly) as the mainstay of induction and maintenance therapy. The use of alternative tyrosine kinase inhibitors such as dasatinib and nilotinib are being investigated in children as well as adults.136 Because there is no clear end point for when or whether any of these tyrosine kinase inhibitors can be safely discontinued, many pediatric oncologists continue to consider hematopoietic stem cell transplantation in remission.
JMML, a myeloproliferative disorder unique to childhood, is characterized by extreme monocytosis, hepatosplenomegaly, thrombocytopenia, and increased fetal hemoglobin.137 Bone marrow morphology is often consistent with myelodysplasia as well as myeloproliferation, and cytogenetics frequently reveals a monosomy 7 clone. Mutations in PTN11, CBL, and other RAS pathway genes have also been noted.138,139 Although the clinical course can be indolent (requiring only intermittent blood product and antibiotic support), JMML patients often progress to frank marrow failure. Neither aggressive AML-type chemotherapy nor splenectomy has been shown to significantly prolong survival, so these approaches are starting to be reserved for those patients who are symptomatic. The most definitive therapy in JMML is an allogeneic BMT.140 Overall DFS with BMT in JMML has been in the 40% to 55% range, with relapse the major reason for failure. Novel therapies targeting the rat sarcoma (RAS), rapidly accelerated fibrosarcoma (RAF), mitogen activated protein kinase (MEK), mammalian target of rapamycin (mTOR), signal transducer and activator of transcription 5 (STAT5), and other signaling pathways are currently under investigation.137
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


