David A. Haake, Paul N. Levett
Leptospira Species (Leptospirosis)
Leptospirosis is a zoonosis of global distribution, caused by infection with pathogenic spirochetes of the genus Leptospira. The disease is greatly underreported, particularly in tropical regions, but recent attempts at surveillance suggest that it may be the most common zoonosis.1 The disease is maintained in nature by chronic renal infection of carrier animals, which excrete the organism in their urine, contaminating the environment. Human infection occurs either by direct contact with infected urine or tissues, or more commonly by indirect exposure to the organisms in damp soil or water. Most human infections are probably asymptomatic; the spectrum of illness is extremely wide, ranging from undifferentiated febrile illness to severe multisystem disease with high mortality rates. The extreme variation in clinical presentation is partly responsible for the significant degree of underdiagnosis.
History
A syndrome of severe multisystem disease, presenting with profound jaundice and renal function impairment, was described by Weil in Heidelberg in 1886. Other descriptions of disease that probably represent leptospirosis were made earlier, but the etiology cannot be definitively ascribed to leptospiral infection.2 Leptospires were first visualized in autopsy specimens from a patient thought to have had yellow fever3 but were not isolated until several years later, almost simultaneously in Germany and Japan.4 Diagnostic confusion between severe icteric leptospirosis and yellow fever continued, with prominent researchers such as Stokes and Noguchi dying in their attempts to discover the etiologic agent.4 Several authoritative reviews have been published.2,4–7
Etiology
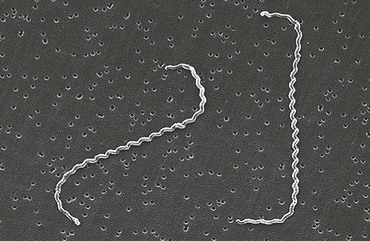
“Leptospira” derives from the Greek leptos (thin) and Latin spira (coiled). Aptly named, the leptospires are a mere 0.1 µm in diameter by 6 to 20 µm in length. The cells have pointed ends, one or both of which is usually bent into a characteristic hook (Fig. 241-1). Motility is conferred by the rotation of two axial flagella underlying the membrane sheath, which are inserted at opposite ends of the cell and extend toward the central region.8 Because of their small diameter, leptospires are best visualized by darkfield microscopy, appearing as actively motile spirochetes (Fig. 241-2). Leptospires are readily cultured in polysorbate-albumin media if specimens are obtained before initiation of antibiotic therapy.9

Historically, the genus Leptospira was classified into two species, L. interrogans and L. biflexa, composed of pathogenic and nonpathogenic strains, respectively. Within each species, large numbers of serovars were differentiated using agglutinating antibodies. Serovar specificity is conferred by lipopolysaccharide (LPS) O-antigens.10 More than 250 serovars of pathogenic leptospires have been described; because of the large number of serovars, antigenically related serovars were grouped into serogroups for convenience in serologic testing.
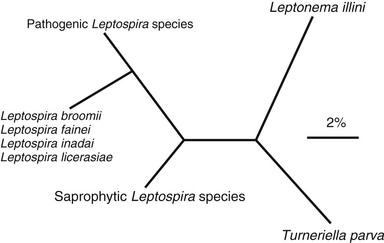
Leptospires are now classified into a number of species defined by their degree of genetic relatedness, determined by DNA reassociation.11,12 There are currently 21 named species, including pathogens (e.g., L. interrogans), nonpathogenic saprophytes (e.g., L. biflexa), and species of indeterminate pathogenicity (e.g., L. inadai) (Table 241-1).13 Some species contain both pathogenic and nonpathogenic strains. This classification is supported by 16S RNA gene sequencing (Fig. 241-3)14 but is quite distinct from the former serologic classification.5
TABLE 241-1
Species of Leptospira and Some Pathogenic Serovars
| SPECIES | SELECTED PATHOGENIC SEROVARS |
| L. interrogans | Icterohaemorrhagiae, Copenhageni, Canicola, Pomona, Australis, Autumnalis, Pyrogenes, Bratislava, Lai |
| L. noguchii | Panama, Pomona |
| L. borgpetersenii | Ballum, Hardjo, Javanica |
| L. santarosai | Bataviae |
| L. kirschneri | Bim, Bulgarica, Grippotyphosa, Cynopteri |
| L. weilii | Celledoni, Sarmin |
| L. alexanderi | Manhao 3 |
| L. alstonii | Sichuan |
| L. meyeri | Sofia |
| L. wolffii | Khorat |
| L. kmetyi | Manilae |
| L. wolbachii | Nonpathogen |
| L. biflexa | Nonpathogen |
| L. vanthielii | Nonpathogen |
| L. terpstrae | Nonpathogen |
| L. yanagawae | Nonpathogen |
| L. idonii | Nonpathogen |
| L. inadai | Indeterminate |
| L. fainei | Indeterminate |
| L. broomii | Indeterminate |
| L. licerasiae | Indeterminate |
The system of serogroup nomenclature has no taxonomic standing but is retained because presumptive serogroup determination by serologic testing has some epidemiologic value. However, serologic responses may bear surprisingly little relationship to the infecting serovar in individual patients.15
The genome sequences of several Leptospira species and strains have been determined,16–20 and sequencing of another 200 strains is under way through the Leptospira Genomics and Human Health Project. The availability of these genome sequences has already led to better understanding of leptospiral pathogenesis.
Epidemiology and Transmission
Leptospirosis is endemic throughout the world. Human infections are endemic in most regions, and the peak incidence occurs in the rainy season in tropical regions and the late summer to early fall in temperate regions.21 In developing countries with poor urban housing standards, leptospirosis outbreaks occur regularly after heavy rainfall and flooding.22 The incidence of leptospirosis is probably grossly underestimated because of limited diagnostic capacity in the regions where the burden of disease is greatest.1 In the United States, the highest incidence is found in Hawaii; active surveillance in 1992 detected an annual incidence of approximately 128 per 100,000.23 Leptospirosis is no longer a nationally notifiable disease in the United States, although it remains notifiable in more than 20 states.
Leptospirosis is maintained in nature by chronic renal infection of carrier animals. The most important reservoirs are rodents and other small mammals, but livestock and companion animals are also significant sources of human infection. Infection of carrier animals usually occurs during infancy, and once infected, animals may excrete leptospires in their urine intermittently or continuously throughout life.
Infection occurs through direct or indirect contact with urine or tissues of infected animals. Direct contact is important in transmission to veterinarians, workers in milking sheds on dairy farms, abattoir workers, butchers, hunters, and animal handlers (transmission has been reported to children handling puppies and to dog handlers). Indirect contact is more common and is responsible for disease following exposure to wet soil or water. The great majority of cases are acquired by this route in the tropics, either through occupational exposure to water as in rice or taro farming or through exposure to damp soil and water during avocational activities.
Recreational exposures have become relatively more important, often in association with adventure tourism to tropical endemic areas. Several large point-source water-borne outbreaks involving attack rates as high as 42% have occurred recently following athletic events.24,25 In recent years there has been an increase in leptospirosis cases among dogs in the eastern regions of North America and in the Midwest,26 associated with a shift in the predominant serovars causing disease.27,28 Veterinary vaccine manufacturers have responded to this problem by licensing new canine vaccines containing these emerging serovars.
Pathogenesis
Leptospires enter the body through cuts and abrasions; mucous membranes or conjunctivae; and aerosol inhalation of microscopic droplets. Swallowing contaminated lake water was the only behavioral risk factor identified in a case-control study of a large leptospirosis outbreak at the 1998 Springfield Triathlon.24 However, the oral mucosae are probably a more important route of entry after ingestion than the intestinal tract. On entering the body, there is widespread hematogenous dissemination, with levels as high as 106 leptospires per milliliter of blood documented by quantitative polymerase chain reaction (PCR).29 Organisms readily penetrate tissue barriers, resulting in invasion of the central nervous system and aqueous humor of the eye. Transendothelial migration of spirochetes is facilitated by a systemic vasculitis, accounting for a broad spectrum of clinical illness. Severe vascular injury can ensue, leading to pulmonary hemorrhage, ischemia of the renal cortex and tubular-epithelial cell necrosis, and destruction of the hepatic architecture resulting in jaundice and liver cell injury with or without necrosis.30
The mechanisms by which leptospires cause disease are not clearly understood. Potential virulence factors include immune mechanisms, toxin production, adhesins, and other surface proteins. Human susceptibility to leptospirosis may be related to poor recognition of leptospiral LPS by the innate immune system.31,32 Human Toll-like receptor (TLR) 4, which responds to extremely low concentrations of gram-negative LPS (endotoxin), appears to be unable to bind leptospiral LPS,32,33 perhaps because of the unique methylated phosphate residue of its lipid A.34 In contrast to human TLR4, mouse TLR4 can recognize leptospiral LPS and has been shown to play an important role in preventing fatal leptospirosis infection in mice.33,35 Direct tissue damage may also be due to production of hemolytic toxins, which may act as sphingomyelinases, phospholipases, or pore-forming proteins.36,37
Immune-mediated mechanisms have been postulated as one factor influencing the severity of symptoms.38 Investigation of the same triathlon outbreak mentioned earlier identified the human leukocyte antigen (HLA) DQ6 as an independent risk factor for leptospirosis.39 The structural location of HLA-DQ6 polymorphisms associated with disease suggested that leptospires produce a superantigen that can cause nonspecific T-cell activation in susceptible individuals. Other immune mechanisms, including circulating immune complexes, anticardiolipin antibodies, and antiplatelet antibodies, have been proposed, but their significance is unproven. Although immune or autoimmune mechanisms may play a role in some cases of leptospiral uveitis,40 a high percentage of such patients have leptospiral DNA in the aqueous humor by PCR.41
Much recent work has focused on the roles of surface lipoproteins in leptospiral pathogenesis.42 The major outer membrane lipoprotein, LipL32, is highly conserved among pathogenic serovars.43 LipL32 is a major target of the human immune response44 and appears to be involved in pathogenesis of tubulointerstitial nephritis.45 Virulent leptospires respond to the increased osmolarity of host tissues by inducing expression of the multifunctional Lig surface proteins that mediate interactions with fibronectin, fibrinogen, and other extracellular matrix factors.46 The Lig proteins are early antigens; IgM antibodies to their immunoglobulin-like repeats develop early in infection, offering an approach to improved detection of acute infection.47 The endostatin-like LenA protein binds the complement regulatory protein, factor H, suggesting an important role in serum resistance.48
Clinical Manifestations
Leptospiral infection is associated with a broad spectrum of severity, ranging from subclinical illness followed by seroconversion to two clinically recognizable syndromes: a self-limited, systemic illness seen in roughly 90% of infections and a severe, potentially fatal illness accompanied by any combination of renal failure, liver failure, and pneumonitis with hemorrhagic diathesis.2,6,7 In some patients, the disease has two distinct phases, an initial septicemic stage that is followed by a temporary decline in fever, followed by an immune phase in which the severe symptoms occur. However, in many severe cases the distinction between these two phases is not apparent; in addition, many patients present only with the onset of the second phase of the illness.
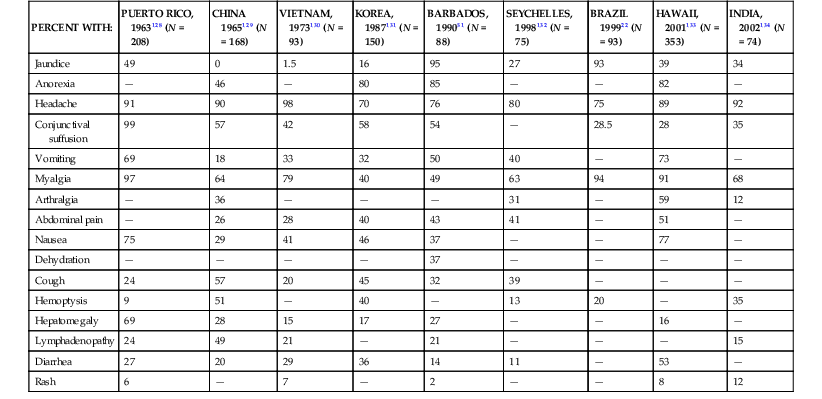
The mean incubation period is 10 days, ranging from 5 to 14 days; determination of precise exposures may be difficult, leading to significant imprecision in estimated incubation times. The acute, septicemic phase of illness begins abruptly with high, remittent fever (38° to 40° C) and headache, chills, rigors, and myalgias; conjunctival suffusion (redness without exudate); abdominal pain; anorexia, nausea, and vomiting; diarrhea; and cough and pharyngitis. A pretibial maculopapular cutaneous eruption occurs rarely (Table 241-2). Conjunctival suffusion and muscle tenderness, most notable in the calf and lumbar areas, are the most characteristic physical findings but may occur in a minority of cases (see Table 241-2). Other less common signs include lymphadenopathy, splenomegaly, and hepatomegaly. The acute phase lasts from 5 to 7 days. Routine laboratory tests are nonspecific but indicative of a bacterial infection. Leptospires can be recovered from blood and cerebrospinal fluid (CSF) during the acute phase of illness, but meningeal signs are not prominent in this phase. Leptospires may also be recovered from urine, beginning about 5 to 7 days after the onset of symptoms (Fig. 241-4). Urinalysis reveals mild proteinuria and pyuria, with or without hematuria and hyaline or granular casts. Death is rare in the acute phase of illness.
TABLE 241-2
Signs and Symptoms on Admission in Patients with Leptospirosis in Large Case Series
| PERCENT WITH: | PUERTO RICO, 1963128 (N = 208) | CHINA 1965129 (N = 168) | VIETNAM, 1973130 (N = 93) | KOREA, 1987131 (N = 150) | BARBADOS, 199051 (N = 88) | SEYCHELLES, 1998132 (N = 75) | BRAZIL 199922 (N = 93) | HAWAII, 2001133 (N = 353) | INDIA, 2002134 (N = 74) |
| Jaundice | 49 | 0 | 1.5 | 16 | 95 | 27 | 93 | 39 | 34 |
| Anorexia | — | 46 | — | 80 | 85 | — | — | 82 | — |
| Headache | 91 | 90 | 98 | 70 | 76 | 80 | 75 | 89 | 92 |
| Conjunctival suffusion | 99 | 57 | 42 | 58 | 54 | — | 28.5 | 28 | 35 |
| Vomiting | 69 | 18 | 33 | 32 | 50 | 40 | — | 73 | — |
| Myalgia | 97 | 64 | 79 | 40 | 49 | 63 | 94 | 91 | 68 |
| Arthralgia | — | 36 | — | — | — | 31 | — | 59 | 12 |
| Abdominal pain | — | 26 | 28 | 40 | 43 | 41 | — | 51 | — |
| Nausea | 75 | 29 | 41 | 46 | 37 | — | — | 77 | — |
| Dehydration | — | — | — | — | 37 | — | — | — | — |
| Cough | 24 | 57 | 20 | 45 | 32 | 39 | — | — | — |
| Hemoptysis | 9 | 51 | — | 40 | — | 13 | 20 | — | 35 |
| Hepatomegaly | 69 | 28 | 15 | 17 | 27 | — | — | 16 | — |
| Lymphadenopathy | 24 | 49 | 21 | — | 21 | — | — | — | 15 |
| Diarrhea | 27 | 20 | 29 | 36 | 14 | 11 | — | 53 | — |
| Rash | 6 | — | 7 | — | 2 | — | — | 8 | 12 |
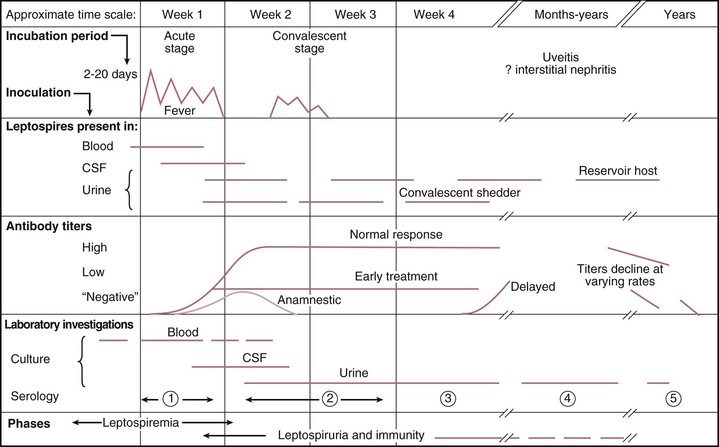
The immune phase of illness generally lasts from 4 to 30 days (see Fig. 241-4). The disappearance of leptospires from the blood and CSF coincides with the appearance of IgM antibodies.7,49 The organisms can be detected in almost all tissues and organs and in urine for several weeks, depending on the severity of the disease. In addition to the acute-phase symptoms described in the preceding paragraph, the immune phase may be characterized by any or all of the following signs and symptoms: jaundice, renal failure, cardiac arrhythmias, pulmonary symptoms, aseptic meningitis, conjunctival suffusion with or without hemorrhage; photophobia; eye pain; muscle tenderness; adenopathy; and hepatosplenomegaly (see Table 241-2). Abdominal pain is not uncommon and may be an indication of pancreatitis.
Aseptic meningitis, with or without symptoms, is characteristic of the immune phase of illness, occurring in up to 80% of cases. In endemic areas, a significant proportion of all aseptic meningitis cases may be caused by leptospiral infection.50 Symptomatic patients present with an intense, bitemporal and frontal throbbing headache with or without delirium. A lymphocytic pleocytosis occurs, with total cell counts generally below 500/mm3. CSF protein levels are modestly elevated between 50 and 100 mg/mL; the CSF glucose concentration is normal. Severe neurologic complications such as meningoencephalitis, hemiplegia, transverse myelitis, or Guillain-Barré syndrome occur only rarely.5
The most distinctive form of severe illness that may develop after the acute phase of illness is Weil’s disease, characterized by impaired hepatic and renal function. More severe cases may progress directly from the acute phase without the characteristic brief improvement in symptoms to a fulminant illness, with fever greater than 40° C and the rapid onset of liver failure, acute renal failure, hemorrhagic pneumonitis, cardiac arrhythmia, or circulatory collapse.7 Mortality rates in patients developing severe disease have ranged from 5% to 40%.2,5,6,51 In a study of 840 hospitalized patients with severe leptospirosis (14% case-fatality rate), risk of death was found to increase with age, especially in adults 40 years of age or older.22,52 Altered mental status has been found to be the strongest predictor of death.22,53 Other poor prognostic signs include acute renal failure (oliguria, hyperkalemia, serum creatinine >3 mg/dL), respiratory insufficiency (dyspnea, pulmonary rales, chest x-ray infiltrates), hypotension, and arrhythmias.22 In jaundiced patients, disturbance of liver function is out of proportion to the rather mild and nonspecific pathologic findings. Conjugated serum bilirubin levels may rise to 80 mg/dL, accompanied by more modest elevations of serum transaminases, alanine aminotransferase, and aspartate aminotransferase, which rarely exceed 200 U/L.54 This is in marked contrast to viral hepatitis. Jaundice is slow to resolve, but death due to liver failure almost never occurs in the absence of renal failure. At autopsy, degenerative changes are seen in hepatocytes, Kupffer cells may be hypertrophied, cholestasis is evident, and erythrophagocytosis and mononuclear cell infiltrates are observed.55 Hepatocellular necrosis is absent.
Kidney involvement is initially characterized by a unique nonoliguric hypokalemic form of renal insufficiency. Hallmarks are impaired sodium reabsorption, increased distal sodium delivery, and potassium wasting. The impairment in sodium reabsorption appears to be due to selective loss of the ENaC sodium channel in the proximal tubular epithelium. The blood urea nitrogen level is usually below 100 mg/dL, and the serum creatinine level is usually below 2 to 8 mg/dL during the acute phase of illness.56 Thrombocytopenia occurs in the absence of disseminated intravascular coagulation and may accompany progressive renal dysfunction.57 Renal biopsy reveals acute interstitial nephritis; immune-complex glomerulonephritis may also be present.58 If electrolyte and volume losses are not replaced, patients progress to oliguric renal failure. In fatal cases, the kidneys are swollen and yellow, with prominent cortical blood vessels.30 Histologic findings include a diffuse, mixed tubulointerstitial inflammatory cell infiltrate of lymphocytes, plasma cells, macrophages, and polymorphonuclear leukocytes, accompanied by focal areas of tubular necrosis.55
Severe pulmonary hemorrhage syndrome (SPHS) can be a prominent manifestation of infection and may occur in the absence of hepatic and renal failure.59 Frank hemoptysis can arise simultaneously with the onset of cough during the acute phase of illness.60 However, hemorrhage is often inapparent until patients are intubated; clinicians should suspect SPHS in patients with signs of respiratory distress whether or not they have hemoptysis. With progressive pulmonary involvement, radiographic abnormalities seen most frequently in the lower lobes evolve from small nodular densities (“snowflake-like”) to patchy alveolar infiltrates; confluent consolidation is uncommon but may occur.61 The pathophysiology of SPHS is consistent with acute respiratory distress syndrome (ARDS) with diffuse lung injury, impaired gas exchange, and hemodynamic changes indicative of septic shock.62 At autopsy, the lungs appear grossly congested and demonstrate focal areas of hemorrhage.55 Histologically, damage to the capillary endothelium leads to congestion with foci of interstitial and intra-alveolar hemorrhage, diffuse alveolar damage, and severe airspace disorganization.63 Inflammatory infiltrates are usually absent.
Congestive heart failure occurs rarely. However, nonspecific electrocardiographic changes are common.64 In more than half of patients receiving continuous cardiac monitoring, cardiac arrhythmias may occur, including atrial fibrillation; flutter and tachycardia; and cardiac irritability, including premature ventricular contractions and ventricular tachycardia.64 Atrial fibrillation is associated with more severe disease.65 Cardiovascular collapse with shock can develop abruptly and in the absence of aggressive supportive care can be fatal. At autopsy, interstitial myocarditis with inflammatory involvement of the conduction system is seen66; acute coronary arteritis and aortitis are also common at postmortem examination.67
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


