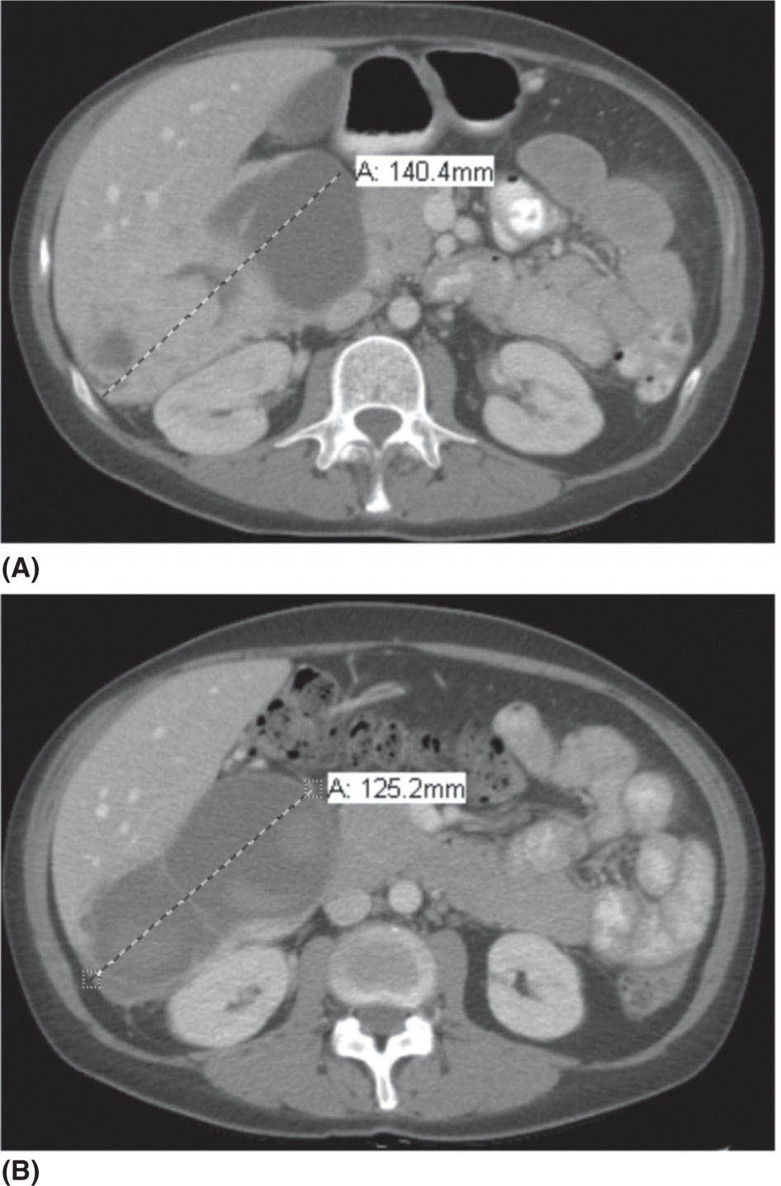
1178 Leiomyosarcoma Leiomyosarcoma (LMS) is a relatively common type of soft tissue sarcoma, commonly with an aggressive clinical course. The two broad categories of LMS include uterine leiomyosarcoma (ULMS) and nonuterine leiomyosarcoma (NULMS), both of which are addressed in this chapter. Accurate diagnosis and pathologic review of LMS are critical, particularly given distinct outcomes based on histology, tumor size, and grade. Multidisciplinary evaluation by a team of pathologists, radiologists, medical oncologists, radiation oncologists, and surgical oncologists is paramount for optimal outcomes. Evaluation is particularly critical given the differences in treatment paradigms for ULMS and NULMS, with unique site-specific considerations for NULMS. Pretreatment consideration for possible LMS is also critical, particularly in patients with uterine leiomyomas, where morcellation procedures are highly discouraged, given possible tumor spillage. histology, leiomyosarcoma, medical oncologists, multidisciplinary evaluation, pathologists, radiation oncologists, radiologists, surgical oncologists, treatment paradigms Histology, Leiomyosarcoma, Oncologists, Pathologists, Radiologists, Therapeutics INTRODUCTION Leiomyosarcoma (LMS) is a malignant mesenchymal tumor originating from the smooth muscle connective tissue. With a propensity for early hematogenous dissemination, LMS, in general, behaves aggressively, with studies showing higher risk for distant recurrence and poorer disease-specific survival than most other sarcoma subtypes. As in other sarcoma subtypes, histologic grade is a fundamental prognostic indicator, and reports show that up to 90% of LMS are moderate to high grade. This translates into survival differences, with a 5-year survival rate for LMS of 40%, which decreases to 10% to 15% for high-grade LMS. This aggressive behavior is paired with a relatively high frequency of LMS cases relative to other soft tissue sarcoma subtypes, with LMS accounting for approximately 10% of all soft tissue sarcomas. There has been a reported decrease in the overall incidence of LMS since 1970; however, this has been attributed most significantly to the reclassification of some previously classified gastric LMS as gastrointestinal stromal tumors with improved immunohistochemical characterization. The two broad categories of LMS include uterine leiomyosarcoma (ULMS) and nonuterine leiomyosarcoma (NULMS), with some unique differences that are also incorporated into the current treatment paradigms. ULMS is associated with higher sensitivity to ifosfamide, greater metastatic potential, and lower median survival of approximately 4.2 years, compared with NULMS, which has a median survival of approximately 8 years.1 Though NULMS can occur in any part of the body with smooth muscle, a common site of origin is the wall of the inferior vena cava in the retroperitoneum. There are also unique molecular aberrations associated with ULMS and NULMS, some of which may influence sensitivity to treatment. Of the three LMS molecular subtypes confirmed by Guo et al. in 2015, types 1 and 2 are linked with extrauterine sites and type 3 is associated with ULMS.2 In addition to the differing molecular aberrations of ULMS and NULMS that are described in further detail in this chapter, LMS has frequent losses in chromosome 10q, which houses the PTEN gene. Loss of PTEN expression ultimately leads to constitutive Akt and mTOR (mammalian target of rapamycin) activation, which has been shown to be important in LMS pathology. Given their distinctive behaviors, ULMS and NULMS are described in greater detail under separate sections, and differences are highlighted when applicable all through the chapter. WORKUP Presenting symptoms for LMS are often nonspecific, typically caused by the primary tumor displacing normal structures, and hence unique to the location. Alternatively, they may present as a palpable mass, such as with an extremity primary or with subcutaneous metastases. Many times, these tumors are incidental discoveries, either on imaging or as a surprise finding on hysterectomy (ULMS). Certain presenting symptoms are unique to their tumor location, with half of ULMS patients with uterine-confined disease presenting with a solitary, large intramural mass that is often palpable. Other presenting symptoms for ULMS may include uterine bleeding, increased abdominal girth, and pelvic pain or pressure. This is in contrast to the LMS of the inferior vena cava, which is often associated with abdominal or flank pain, with other possible presenting symptoms including abdominal distention, Budd–Chiari syndrome, lower extremity edema, and even deep venous thrombosis. Initial workup includes imaging approaches to visualize the primary tumor and possible sites of metastatic disease. With regard to primary tumor imaging, typical approaches involve use of MRI with and without contrast for extremity soft tissue tumors and CT with contrast for chest, abdominal, or pelvic masses. Chest and abdominal CT scan is required in the initial workup, given the frequent 118hematogenous spread seen in LMS, with liver and lung being the common sites of metastatic disease. Other sites of metastatic disease include subcutaneous sites, bone, central nervous system, and rarely, the gastrointestinal system. Morphologic imaging features associated with LMS depend on imaging modality and site of the primary tumor. On CT imaging, LMS is typically heterogeneous with central low attenuation that represents necrosis. Calcification is typically not observed in LMS. MRI shows signal characteristics including T1 isointense to muscle, T2 fat-suppressed predominantly hyperintense, and T2 non–fat-suppressed intermediate to hypointense to surrounding fat. MRI may also show cystic foci within the tumor. As with other soft tissue sarcomas, there are no pathognomonic findings for LMS on imaging, which is why the diagnostic approach for LMS involves pretreatment biopsy with appropriate pathologic review. LMS is typically reactive for smooth muscle actin, h-caldesmon, and desmin on immunohistochemistry; however, none of these markers are specific for smooth muscle differentiation. Pathologic review must also include histologic grading, which, as mentioned above, is an independent indicator of disease-specific survival as well as probability of metastatic disease. TREATMENT OF LOCALIZED DISEASE Nuances specific to the treatment of ULMS and NULMS are detailed in their respective sections; however, for both ULMS and NULMS, the cornerstone of treatment is complete surgical excision with wide negative margins, whenever possible. Large NULMS tumors in the retroperitoneum are associated with anatomic constraints that often make complete surgical resection challenging. R0 resection remains the most important prognostic factor associated with survival. Use of neoadjuvant or adjuvant radiation and chemotherapy should be considered based on unique clinical characteristics similar to other soft tissue sarcomas. Given the high propensity for metastases with high-grade LMS and their relative sensitivity to chemotherapy, it is common clinical practice to consider neoadjuvant or adjuvant chemotherapy in large tumors >5 cm in size, similar to other soft tissue sarcomas.3 Neoadjuvant/adjuvant chemotherapy use in ULMS is even less standardized, and the approach varies among different centers and whether patients are managed by sarcoma oncologists versus gynecologic oncologists. This is discussed in further detail in the section “Uterine Leiomyosarcoma.” Preferred chemotherapy regimens for neoadjuvant or adjuvant treatment differ by LMS subtype with doxorubicin plus dacarbazine used in NULMS and either doxorubicin plus ifosfamide or gemcitabine plus docetaxel used in ULMS. ULMS has no clear indication for adjuvant radiation, but can be discussed on a case-by-case basis if there is a concern for positive margin surgery or for a localized pelvic recurrence. Neoadjuvant radiation is given as a lower dose than adjuvant radiation, and thus is generally preferred to minimize toxicity to the bowel and other surrounding structures that occupy the resection cavity. For NULMS, radiation has been shown to improve local control with decreased local recurrence rates, but without a proven overall survival benefit. This specifically applies to the extremity and trunk location, where neoadjuvant/adjuvant radiation therapy is standard for intermediate to high-grade, ≥5 cm soft tissue sarcomas.4 There is a paucity of data regarding its use for primary retroperitoneal soft tissue sarcomas. Ultimately, preoperative or postoperative radiation is the standard of care for LMS patients with intermediate or high-grade, large tumors involving the limbs and trunk, whereas it is not standard for retroperitoneal NULMS or ULMS. FOLLOW-UP AFTER DEFINITIVE TREATMENT Following definitive treatment of initial localized disease, surveillance recommendations are similar to other soft tissue sarcomas with history and physical exams taken every 3 to 6 months for 2 to 3 years, which is then lengthened to every 6 months for the next 2 years and then annually. Surveillance imaging is incorporated into these visits to include the primary site and chest imaging. For retroperitoneal or pelvic tumors, imaging should include the entire abdomen and pelvis. The imaging schedule can be altered based on the estimated risk of locoregional recurrence. TREATMENT OF METASTATIC DISEASE As previously noted, LMS is a highly aggressive tumor with a propensity for early hematogenous dissemination. Common sites of metastases in both ULMS and NULMS are lung and liver. Liver is a 119common metastatic site for visceral primaries, whereas lung is the most common site of metastasis in patients with ULMS and NULMS with extremity primary tumors. In addition to this high risk for metastatic disease, LMS is associated with a high risk of relapse. Both relapsed and metastatic disease are associated with a high mortality rate. Treatment intent in metastatic LMS is palliative, with the goals of improving symptoms and quality of life, decreasing the tumor bulk, and improving survival. Similar to other soft tissue sarcomas, first-line chemotherapy involves anthracycline-based regimens, but the most data are available for doxorubicin-based therapies. Combination chemotherapy with doxorubicin, using either doxorubicin plus ifosfamide or doxorubicin plus dacarbazine, has historically shown improved response rates, but not improved overall survival. Given this, these doxorubicin-based combination therapies are typically utilized when the response is critical, such as in symptomatic disease, potentially resectable disease, or when the tumor threatens the end organ function. The combination of doxorubicin plus olaratumab uniquely did not improve the response rate compared to single-agent doxorubicin, but did improve overall survival in a Phase 2 study. The Phase 3 results of the doxorubicin plus olaratumab combination did not confirm the findings of the Phase 2 study; thus, doxorubicin plus either ifosfamide or dacarbazine remains standard. Uniquely, ULMS has been associated with higher response to ifosfamide, whereas it has limited activity as a single agent in NULMS, suggesting that doxorubicin plus ifosfamide may be a more appropriate frontline regimen for ULMS as compared with NULMS. In NULMS, given the poorer response to ifosfamide, frontline chemotherapy with doxorubicin doublets is now typically doxorubicin plus dacarbazine. Beyond the first-line treatment with anthracyclines, other active cytotoxic agents for metastatic LMS include gemcitabine with or without docetaxel, pazopanib, and trabectedin. If not given in the first-line setting, dacarbazine (or temozolamide) and ifosfamide (particular to ULMS) also has activity. Gemcitabine plus docetaxel has a higher activity in LMS (50% response rate) compared to most other soft tissue sarcoma subtypes, and hence is often used in the frontline setting for LMS.5 There has been conflicting evidence regarding the benefit of addition of docetaxel to single-agent gemcitabine, with one trial showing improved efficacy of the combination in the second-line treatment of LMS,5 but a second randomized trial specific to LMS patients showing no improvement in response rate, progression-free survival, and overall survival in patients treated with gemcitabine plus docetaxel as compared with gemcitabine alone.6 In healthy patients with a good performance status, the use of gemcitabine plus docetaxel is reasonable; however, gemcitabine single agent is a reasonable treatment strategy, particularly for those patients with poorer performance status or intolerance to the more toxic side effects associated with the gemcitabine plus docetaxel combination. In further lines of therapy, the multikinase inhibitor pazopanib was approved for treatment of soft tissue sarcomas, including LMS, that have progressed after prior cytotoxic therapy, with improved progression-free survival but not overall survival found in the Phase 3 trial as compared with a control group. The cytotoxic agent trabectedin is also approved for LMS and liposarcomas and should be considered in later lines of therapy. As with other therapies, tumor shrinkage with trabectedin is often preceded by early tissue changes. This can lead to radiographic response in the form of decreasing density, as seen in Figure 8.1, which may not be captured if utilizing only Response Evaluation Criteria in Solid Tumors (RECIST) criteria for response assessments and is an important consideration when assessing individual patients as well as designing clinical trials. Beyond the aforementioned treatments, the Phase 2 REGOSARC trial showed improved progression-free survival with regorafenib in patients with nonadipocytic soft tissue sarcomas including LMS. Additionally, while only approved in liposarcoma, a randomized Phase 3 trial of eribulin versus dacarbazine showed some disease stabilization with eribulin in LMS, but not superior to dacarbazine, suggesting this as a treatment option in later lines of therapy for LMS patients. Various options are under investigation for LMS, including a multitarget tyrosine kinase inhibitor anlotinib. In the Phase 2 clinical trial of anlotinib in soft tissue sarcoma patients, reported outcomes showed that almost 70% of LMS patients were progression free at 12 weeks with a median progression-free survival of 11.07 months in LMS patients. There is an ongoing randomized study of dacarbazine versus anlotinib in LMS to confirm this activity. Additionally, while not considered the standard of care, mTOR inhibitors can be considered in LMS patients, given the loss of PTEN expression in these tumors, leading to Akt and mTOR activation, which is thought to be instrumental in LMS. In a Phase 2 clinical trial in advanced bone and soft tissue sarcomas, ridaforolimus, a second-generation mTOR inhibitor, showed a median progression-free survival of 15.3 weeks and a higher clinical benefit rate in LMS than other cohorts (33.3%). More recently, immunotherapy combinations have been evaluated, as they were felt to have potential in LMS, given the presence of PD-1 (programmed cell death 1 protein) expression on some tumors, high 120mutation rates, and high levels of T-cell–related gene expression. While these findings would suggest immunogenicity, the SARC028 trial notably showed no objective responses in patients with LMS treated with single-agent pembrolizumab. Additionally, an early Phase 2 trial of nivolimumab in patients with metastatic ULMS also failed to show a response. This suggests that immunotherapy combinations are likely necessary to overcome possible reasons for poor single-agent immunotherapy response, including immunosuppressive tumor-activated macrophages. To this end, Alliance A091401 did show responses in ULMS (n = 1) and NULMS (n = 1) in the group receiving the nivolumab plus ipilimumab combination. Additionally, molecular analysis has suggested that PTEN loss in LMS may lead to resistance to PD1 inhibitors, which may need to be addressed in future immunotherapy treatment paradigms for LMS patients. FIGURE 8.1 (A) Pretreatment imaging showing a pathologically confirmed leiomyosarcoma (peritoneal and liver metastases). (B) Imaging following just two cycles of doxorubicin and dacarbazine showed tumor response with increased cystic changes, highlighting that initial radiographic response can be in the form of decreasing density that may not always fulfill the RECIST response criteria. RECIST, Response Evaluation Criteria in Solid Tumors.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


