Langerhans Cell Histiocytosis
Suman Malempati
H. Stacy Nicholson
Langerhans cell histiocytosis (LCH) is a disorder characterized by clonal proliferation of cells in the mononuclear phagocyte system. Since the first case was described more than a century ago,1 LCH has often been a source of confusion, perhaps best demonstrated by the several labels given to the disorder during the past 100 years. Since 1985, Langerhans cell histiocytosis has been the preferred term,2, 3 replacing histiocytosis X, coined in 1953.4 The X demonstrated the lack of knowledge about the etiology and pathophysiology of LCH, and about how the different clinical syndromes were related. The term histiocytosis X did serve to bind the syndromes, which included Hand-Schuller-Christian syndrome, Letterer-Siwe disease, eosinophilic granuloma, Hashimoto-Pritzker syndrome,5 self-healing histiocytosis,6 and pure cutaneous histiocytosis,7 into one clinical entity. The term Langerhans cell histiocytosis reflects the central role of the Langerhans cell in these diseases. LCH also distinguishes these disorders from other histiocytic syndromes, which include primary and secondary hemophagocytic lymphohistiocytosis, Rosai-Dorfman disease, and neoplastic disorders such as acute monocytic leukemia, malignant histiocytosis, and true histiocytic lymphoma.3 This chapter focuses on LCH.
HISTORY
The clinical triad of defects in membranous bone, exophthalmos, and polyuria in children, which became known as Hand-Schuller-Christian disease,8 was described in 1921 in a review by Hand.9 Hand described the first six reported cases, including those of Christian, Schuller, and his own first case reported in 1893.1 All six patients had hepatosplenomegaly, lymphadenopathy, and bone lesions. However, not all had exophthalmos and polyuria. Attempts to link the disorder to xanthoma tuberosum, lipid storage disease, and the xanthomatoses proved unsuccessful, and to this day, no evidence exists that a specific biochemical defect is responsible for the condition. However, the foamy or xanthoma cell came to be regarded as a pathognomonic feature of the syndrome.10
A different syndrome, consisting of fever, bilateral otitis media, hepatosplenomegaly, and adenopathy in a young infant who died, was described by Letterer in 1924.11 In 1933, Siwe described a 16-month-old girl who died after a 3-month illness characterized by fever, hepatosplenomegaly, lymphadenopathy, neutrophilia, and a destructive lesion in her fibula.12 At autopsy, massive infiltrates of large cells resembling histiocytes were found. Siwe reviewed five other cases from the literature (including that of Letterer) and concluded that they constituted a single clinical entity.12
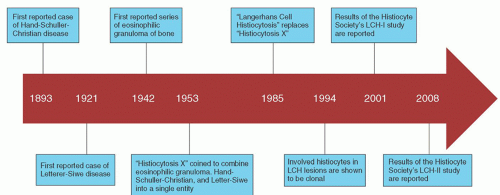 FIGURE 60.1. Historical timeline demonstrating the major discoveries regarding the pathogenesis and treatment of Langerhans cell histiocytosis (LCH). |
In 1940, two groups of investigators described a syndrome in which a characteristic feature was infiltration of bone by eosinophilic granulomas.13, 14 In 1942, Green and Farber described a series of patients with eosinophilic granulomas of bone, which usually healed promptly after irradiation or curettage.15 They noted that these cases shared pathologic features similar to those of the Letterer-Siwe and Hand-Schuller-Christian syndromes. In 1953, because of the similarity of the histiocytes observed in these three disorders, Lichtenstein combined them into a single entity called histiocytosis X to indicate their unknown cause.4 The recognition that these three disorders were related was an important contribution. The term Langerhans cell histiocytosis, proposed in 1985, reflects an improved understanding of these disorders.3
Despite our understanding of the central role played by the Langerhans cells in LCH, little is known about the etiology of LCH. Although clinical features of LCH were first recognized over a century ago, the pathogenesis remains uncertain. In 1994, two separate groups detected clonality of the involved histiocytes from LCH lesions.16, 17 Clonality was demonstrated in LCH cells from unifocal lesions as well as disseminated disease. However, debate continues as to whether LCH represents a neoplastic disorder or a reactive proliferation of histiocytes (Fig. 60.1).
EPIDEMIOLOGY
Although estimates vary by region and time period, the annual childhood incidence of LCH has been estimated to be approximately 4 to 9 cases per million.18, 19, 25 The discrepancy between epidemiologic studies is likely due to variation in reporting and data collection. Across studies, there is consistently a slight
predominance of cases in males.18, 19, 20 LCH occurs less frequently in adults with an incidence of 1 to 2 cases per million per year.21 The disease is more common and tends to be more severe in younger children. For children under 1 year of age, the annual incidence is 9.9 cases per million.22 Almost all cases of multisystem LCH occur before 5 years of age.23, 24, 25 In an exploratory case-control study comparing 177 children with LCH to children with cancer and community controls,20 LCH was associated with a family history of benign tumors and less strongly with feeding problems during infancy. Other factors associated with LCH in this study were maternal urinary tract infections during pregnancy and blood transfusions during infancy. Factors not associated with LCH were the typical childhood viral infections and medication use. In a case-control study of 459 children with LCH, which compared risk factors in children with LCH to those in both community and cancer controls,26 LCH was associated with neonatal infections, solvent exposure, and thyroid disease in the proband or the family. Childhood immunizations appeared to be protective. Reports of seasonal variation in incidence are conflicting18, 25 Familial clustering of LCH has been observed, suggesting that a genetic predisposition may exist.27 The concordance rates between dizygotic and monozygotic twins are 33% and 80%, respectively. In addition, patients with LCH may have a predisposition to cancer and vice versa. Links with both solid tumors and leukemia have been documented.28 Several cases of LCH developing in patients with acute lymphoblastic and acute myelogenous leukemia have been reported.29, 30, 31, 32 Patients with LCH have genetic instability and increased chromosomal breakage, which also suggests a genetic predisposition to malignancy.33, 34, 35
predominance of cases in males.18, 19, 20 LCH occurs less frequently in adults with an incidence of 1 to 2 cases per million per year.21 The disease is more common and tends to be more severe in younger children. For children under 1 year of age, the annual incidence is 9.9 cases per million.22 Almost all cases of multisystem LCH occur before 5 years of age.23, 24, 25 In an exploratory case-control study comparing 177 children with LCH to children with cancer and community controls,20 LCH was associated with a family history of benign tumors and less strongly with feeding problems during infancy. Other factors associated with LCH in this study were maternal urinary tract infections during pregnancy and blood transfusions during infancy. Factors not associated with LCH were the typical childhood viral infections and medication use. In a case-control study of 459 children with LCH, which compared risk factors in children with LCH to those in both community and cancer controls,26 LCH was associated with neonatal infections, solvent exposure, and thyroid disease in the proband or the family. Childhood immunizations appeared to be protective. Reports of seasonal variation in incidence are conflicting18, 25 Familial clustering of LCH has been observed, suggesting that a genetic predisposition may exist.27 The concordance rates between dizygotic and monozygotic twins are 33% and 80%, respectively. In addition, patients with LCH may have a predisposition to cancer and vice versa. Links with both solid tumors and leukemia have been documented.28 Several cases of LCH developing in patients with acute lymphoblastic and acute myelogenous leukemia have been reported.29, 30, 31, 32 Patients with LCH have genetic instability and increased chromosomal breakage, which also suggests a genetic predisposition to malignancy.33, 34, 35
PATHOLOGY AND PATHOPHYSIOLOGY
The basic histologic lesion in LCH is granulomatous, with lesions containing histiocytes, mature eosinophils, and lymphocytes.36 Other cells present may include giant cells, neutrophils, and plasma cells. Initially, lesions are proliferative and dominated by histiocytes, some of which are Langerhans cells. Although mitotic figures may occasionally be identified, the histiocytes are not neoplastic by histologic criteria. As lesions progress, necrosis may develop, and the number of eosinophils and phagocytic cells containing cellular debris increases. Ultimately, xanthomatous changes and fibrosis may occur, and late in the course of disease, Langerhans cells may no longer be demonstrable. Multinucleated giant cells occasionally are prominent, especially in bone and lymph nodes. The histologic findings do not correlate with the extent or aggressiveness of disease.37
The Langerhans cell is the sine qua non of the diagnostic lesion. Langerhans cells are dendritic antigen-presenting cells that are normally found in skin and other organs. Langerhans cells are classified as dendritic cells because of their capacity to form long cytoplasmic extensions through which they establish intimate contact with other cells. The presence of fascin, a highly selective marker of dendritic cells, on the surface of Langerhans cells confirms their derivation from dendritic cells.38 Langerhans cells are found primarily in normal epidermis, but also are evident in lymph nodes and spleen. A hematopoietic progenitor of Langerhans cells has been identified in normal bone marrow.39 In the skin, Langerhans cells form a trap for external contact antigens and are involved in delayed hypersensitivity. Despite low phagocytic activity, they fix antigens for presentation to other cells, especially T lymphocytes.40
Although its demonstration is essential for diagnosis, the Langerhans cell may constitute no more than a small proportion of histiocytes within lesions. Viewed by light microscopy, these cells appear as large mononuclear cells with few cytoplasmic vacuoles and little or no phagocytic material.41 The nuclei are irregularly shaped and contain a fine chromatin pattern. Electron microscopic or immunohistochemical studies may be required to identify Langerhans cells with confidence. The former demonstrate structures known as Birbeck granules (Langerhans bodies, X granules),42 rod-shaped organelles with a central striation and occasional terminal vesicular dilation, giving them a tennis racket appearance (Fig. 60.2). Birbeck granules are thought to be produced by invagination of the cell membrane, and their function is not known. The other feature that conclusively establishes the diagnosis of LCH is the demonstration of either CD1a antigen or CD207 (Langerin) on the surface of LCH cells by immunohistochemical staining.36, 43 Langerin is a cell-surface receptor that induces the formation of Birbeck granules.44 Langerin expression has recently been recognized as a sensitive and relatively specific marker in establishing the diagnosis of LCH. Other distinctive features of Langerhans cells that can be demonstrated with immunohistochemical techniques include the expression of S-100 protein,41 Ia-like antigen,45 and CD101.46
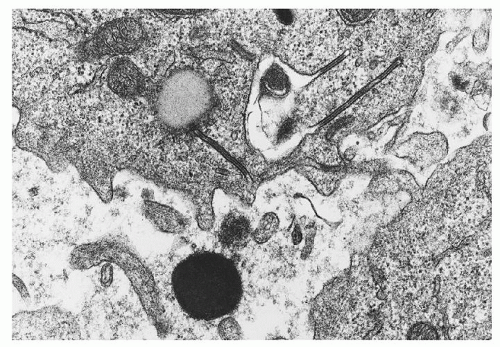 FIGURE 60.2. Electron micrograph of Langerhans cells from a bone lesion demonstrating characteristic Birbeck granules with a trilaminar structure. (Magnification ×45,000; courtesy of Roma Chandra.) |
The intralesional histiocytes of LCH were previously thought to be similar to normal Langerhans cells found in skin. However, it is now recognized that pathologic Langerhans cells or LCH cells are a less differentiated and more activated type of dendritic cell.47 CD1a-positive LCH cells from bone lesions express CD14 and CD68, monocyte antigens that are not expressed or are expressed at low levels in normal skin Langerhans cells.48 A recent study demonstrated a gene expression profile of intralesional LCH cells that was distinct from that of normal skin Langerhans cells.49 LCH cells also express the immature dendritic cell marker CCR6 in the absence of CCR7, which is expressed on mature dendritic cells.50 LCH cells exhibit a degree of migration capability, which is absent in mature dendritic cells. LCH cells can be induced to differentiate in the presence of CD40 ligand in vitro.48 However, as CD40 ligand is abundant in LCH lesions, the reason for maturational arrest is unclear.51 Abnormal cellular adhesion molecules in LCH cells, suggested by the presence of CD2, CD11a, CD11b, and CD11d, may contribute to the migration of Langerhans cells into LCH lesions, as well as their abnormal persistence and proliferation.52 Compared to normal Langerhans cells, LCH cells are defective in their alloantigen-presenting activity.53
The focal accumulation of Langerhans cells, macrophages, lymphocytes, and eosinophils suggests that LCH is immunologically mediated. This reasoning is supported by both histologic abnormalities of the thymus and disturbances of immunoregulation in patients with active disease.54 Thymic abnormalities were noted by Letterer in his initial description of the disorder, later known as Letterer-Siwe disease.11 Abnormalities noted on pre-therapy biopsy samples and postmortem materials include dysmorphic
changes, dysplasia, and nonspecific involution.55, 56, 57 In patients with LCH, the number of normal thymocytes (expressing CD6) and late differentiating suppressor lymphocytes (expressing CD8) is decreased.58 These abnormalities are noted even when the disease is limited in its distribution.
changes, dysplasia, and nonspecific involution.55, 56, 57 In patients with LCH, the number of normal thymocytes (expressing CD6) and late differentiating suppressor lymphocytes (expressing CD8) is decreased.58 These abnormalities are noted even when the disease is limited in its distribution.
Recognition of the possible pathogenic significance of thymic abnormalities prompted several studies of T lymphocytes in blood. The demonstration of decreased numbers of H2 receptors on blood lymphocytes suggested loss of T-suppressor cells.59 This loss was confirmed by quantitation of T-cell subsets in patients with active disease. Both the relative and absolute numbers of suppressor T lymphocytes (CD8+ cells) are decreased, resulting in an increase in the T4-to-T8 ratio. Suppressor cell activity as measured by the concanavalin A and indomethacin stimulation assays also is poor.59 That T-suppressor cell deficiency may be causally related to a functionally abnormal thymus is suggested by the normalization of T4-to-T8 ratios with thymic extract.57 Moreover, the apparent ability of crude thymic extract to reverse disease activity in some patients suggests that the T-cell abnormalities may be of primary pathogenic significance.57
Although less consistent, other abnormalities in immune regulation also have been described, including hypergammaglobulinemia,60 deficiency in antibody-dependent monocyte-mediated cytotoxicity,61 and abnormal in vitro response to mitogens and antigens.55 However, most investigators note normal mitogeninduced responses and normal delayed hypersensitivity.62 Also, several cytokines are increased in LCH.63, 64 Cytokines serve as mediators of inflammation, regulators of lymphocyte growth and differentiation, and activators of specialized effector cells. In tissue culture, Langerhans cells purified from bony LCH lesions secrete interleukin-1 (IL-1) and prostaglandin E2, both of which induce bone resorption in vitro.65 These lesions also secrete angiotensinconverting enzyme, transforming growth factor-β1 and IL-2.66 These mediators are probably responsible for the osteolytic lesions that are a prominent clinical feature of LCH. Other cytokines that have been demonstrated to be increased in the serum of LCH patients include granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-3, IL-8, IL-10, and tumor necrosis factor-α.67 These cytokines are related to local activation of T lymphocytes and other inflammatory leukocytes. In addition, GM-CSF receptors are expressed by Langerhans cells.68 GM-CSF induces Langerhans cell proliferation and activation in vitro, and serum levels of GM-CSF have been correlated with the extent of disease.68
The link between proliferation of Langerhans cells and immune dysfunction has not been worked out entirely. One suggestion is that the disorder results from a physiologically appropriate response of the Langerhans cell to an external antigen or neoantigen, possibly infectious in origin. However, the lack of seasonal variation or geographic clustering argues against an infectious basis. In addition, no direct evidence for a viral etiology (viral particles or nuclear material) in LCH lesions has been demonstrated.69, 70 Alternatively, LCH may result from an appropriate response of the Langerhans cell to abnormal signals from other cells in the immune system, perhaps from T lymphocytes. In either event, deficiency of T-suppressor cells may disrupt the mechanism for termination of immune responses. Failure of this homeostatic mechanism could result in unrestrained macrophage proliferation. Patients with interferon-γ deficiency present with findings that mimic LCH.71
TABLE 60.1 SYSTEMS AFFECTED ON DIAGNOSIS OF LANGERHANS CELL HISTIOCYTOSIS BY AGE | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Whether LCH is primarily a reactive or neoplastic process has long been debated. The predominant theory has been that LCH is a reactive process to an unknown stimulus.72 However, in 1994 two groups showed that the lesional cells are clonal16, 17; clonality occurred in patients with all forms of LCH, including acute disseminated LCH and unifocal LCH, and in those with intermediate forms of the disease. T-cells within LCH lesions were polyclonal.16 Interestingly, LCH occurs in some individuals with malignant disorders.28 Most cases of malignancy occur after treatment for LCH, although acute lymphoblastic leukemia, in particular, may precede the diagnosis of LCH.31, 32 The presence of chromosomal instability and mutational events reported in a few studies support a neoplastic process.34, 35 However, at least some cases of pulmonary LCH are not clonal.73 Moreover, the immature phenotype of LCH cells appears dependent on the microenvironment, suggesting a reactive process. Therefore, although the clonality of LCH cells has fueled the argument that LCH may be a neoplastic disorder, the debate continues.
CLINICAL FEATURES
LCH can present along a continuum of illness, ranging from indolent to explosive disease. In some patients, pathologic lesions are solitary, whereas in others they are widely disseminated. Moreover, the distribution of lesions in a given patient may vary considerably over time. Although LCH can occur at any age, it occurs with greatest frequency in infants and children. The median age at diagnosis for all disease variants is 3 to 6 years.18, 19 The acute disseminated form of the disease characteristically occurs in younger children and almost all cases occur before the age of 5 years.25 Children younger than 1 year old, in particular, often present with multiple organ involvement.22, 74 The more indolent forms of LCH occur primarily in older children and young adults.75 Approximately 70% of cases of LCH in children involve a single organ system, with bone being the most common site.18, 19, 22, 25 Table 60.1 shows the distribution of involved sites
by age at diagnosis of LCH. The most commonly involved organ in adults is bone, often accompanied by an adjacent soft tissue mass.76, 77 Other organs that are involved less often in adults include the lungs and pituitary gland.76 In adults, multisystem disease, including liver, lymph node, and bone marrow involvement, is extremely rare.
by age at diagnosis of LCH. The most commonly involved organ in adults is bone, often accompanied by an adjacent soft tissue mass.76, 77 Other organs that are involved less often in adults include the lungs and pituitary gland.76 In adults, multisystem disease, including liver, lymph node, and bone marrow involvement, is extremely rare.
The traditional classification of clinical variants was based on patterns of organ involvement.8 Eosinophilic granuloma was used to describe a syndrome characterized by single or multiple bone lesions in the absence of visceral involvement.13 When granulomas involved the liver, spleen, lymph nodes, skin, central nervous system (CNS), or bone marrow as well as bones, the disorder was called Letterer-Siwe disease.11, 12 The triad of multiple bone lesions, exophthalmos (resulting from retro-orbital granulomas), and diabetes insipidus (DI; the result of hypothalamic or pituitary involvement) constituted Hand-Schuller-Christian disease (Figs. 60.3 and 60.4).9 The separation of eosinophilic granuloma of bone from syndromes characterized by visceral dissemination proved to be useful prognostically. However, the distinction between Letterer-Siwe disease and Hand-Schuller-Christian disease was often subtle and clinically irrelevant. The current classification is based on the number of organ systems involved and the number of sites involved within an organ system.23, 78 The main classifications of disease are unifocal eosinophilic granuloma (i.e., single-system, single-site disease, usually in bone), multifocal eosinophilic granuloma (i.e., single-system, multiplesite disease, usually in bone), and acute disseminated histiocytosis (i.e., multisystem disease). The presence and degree of organ dysfunction are important distinctions in those with multisystem disease.78
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


