- Regulation of fuel homeostasis in mammals is dependent on numerous small endocrine organs known as islets of Langerhans, which are located in the pancreas.
- Islets contain β-cells which are the only source of the polypeptide hormone insulin.
- β-Cells are equipped to detect changes in circulating nutrients.
- β-Cells respond to elevated levels of nutrients by initiating an insulin secretory response which will induce the storage of circulating nutrients in liver, muscle and adipose tissue.
- β-Cells can also respond to a wide range of other signals including hormones, neurotransmitters and neuropeptides, which modify the insulin secretory response to circulating nutrients.
- Complex interactions between islet cells, the autonomic nervous system and gastrointestinal incretin hormones allow precise integration between metabolic fuel intake, usage and storage.
Introduction
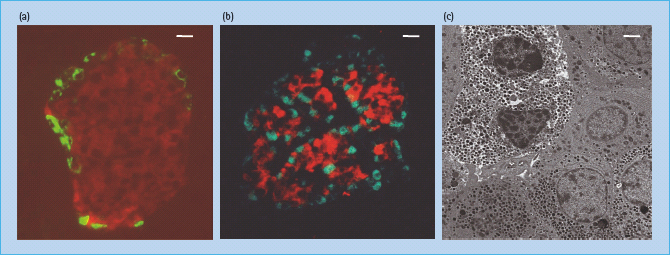
The German anatomy student Paul Langerhans first described in 1869 the “islands of clear cells” distributed throughout the pancreas [1] but he did not realize the physiologic significance of these cell clusters, which are today known as islets of Langerhans. We now know that islets are the endocrine compartment of the pancreas, comprising around 2–3% of the total pancreatic volume. Islets are approximately spherical (Figure 6.1a), with an average diameter of 100–200μm, and a healthy human pancreas may contain up to a million individual islets, each having its own complex anatomy, blood supply and innervation.
Figure 6.1 Anatomy of the islet of Langerhans. (a) Mouse islet. The figure shows a section through a mouse pancreas in which insulin and glucagon are identified by red and green immunofluorescence, respectively, demonstrating the typical β-cell core surrounded by a thin mantle of α-cells. In mouse islets β-cells comprise approximately 80% of the endocrine cell mass. Scale bar is 10 μm. Image courtesy of V. Foot, King’s College London. (b) Human islet. The figure shows a section through a human pancreas in which insulin and glucagon are identified by red and green immunofluorescence respectively, demonstrating the less organized structure of the human islet when compared with mouse islets. In human islets β-cells comprise approximately 50–60% of the endocrine cell mass. Scale bar is 10 μm. Image courtesy of V. Foot, King’s College London. (c) Transmission electron micrograph of human islet cells. The figure shows a transmission electron micrograph of several cells within a human islet. The two cells in the top left with the electron dense secretory granules surrounded by a clear halo are β-cells. The cells in the lower half of the micrograph are α-cells. Scale bar is 2 μm. Data from Jones & Persaud, unpublished.
Islet structure and function
Islet anatomy
A typical mammalian islet comprises several thousand endocrine cells including the insulin-expressing β-cells (∼60% of adult human islet cells), glucagon-expressing α-cells (20–30%), somatostatin-expressing δ-cells (∼10%), pancreatic polypeptide-expressing cells (<5%), and ghrelin-expressing cells (∼1%). The anatomic arrangement of islet cells varies between species. In rodents, the majority β-cell population forms a central core surrounded by a mantle of α-cells and δ-cells (Figure 6.1a), but human islets show less well-defined organization with α-cells and δ-cells also being located throughout the islet (Figure 6.1b) [2].
Islets are highly vascularized, and receive up to 15% of the pancreatic blood supply despite accounting for only 2–3% of the total pancreatic mass. Each islet is served by an arteriolar blood supply that penetrates the mantle to form a capillary bed in the islet core. Earlier studies using vascular casts of rodent islets suggested that the major route of blood flow through an islet was from the inner β-cells to the outer α and δ-cells [3], but more recent studies using optical imaging of fluorescent markers to follow islet blood flow in vivo [4] reveal more complex patterns of both inner-to-outer and top-to-bottom blood flow through the rodent islet.
Islets are well supplied by autonomic nerve fibers and terminals containing the classic neurotransmitters acetylcholine and norepinephrine, along with a variety of biologically active neuropeptides [5]. Vasoactive intestinal polypeptide (VIP) and pituitary adenylate cyclase-activating polypeptide (PACAP) are localized with acetylcholine to parasympathetic nerves, where they may be involved in mediating prandial insulin secretion and the α-cell response to hypoglycemia [6]. Other neuropeptides, such as galanin and neuropeptide Y (NPY), are found with norepinephrine in sympathetic nerves where they may have a role in the sympathetic inhibition of insulin secretion, although there are marked inter-species differences in the expression of these neuropeptides [5].
Intra-islet interactions
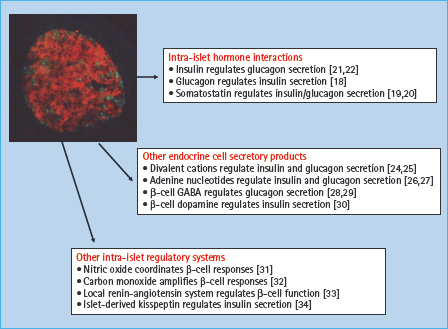
The anatomic organization of the islet has a profound influence on the ability of the β-cells to recognize and respond to physiologic signals [7–9]. There are a number of mechanisms through which islet cells can communicate, although the relative importance of the different mechanisms is still uncertain [10]. Islet cells are functionally coupled through a network of gap junctions, and gene deletion studies in mice have highlighted the importance of gap-junctional coupling via connexin 36 in the regulation of insulin secretory responses [11,12]. Cell–cell contact through cell surface adhesion molecules offers an alternative communication mechanism, and interactions mediated by E-cadherin [13,14] or ephrins [15] have been implicated in the regulation of β-cell function. A further level of control can be exerted via intra-islet paracrine and autocrine effects in which a biologically active substance released by one islet cell can influence the functional status of a neighboring cell (paracrine), or of itself (autocrine). Figure 6.2 shows some of the molecules that have been implicated in this type of intra-islet cell–cell communication. Thus, islet cells can interact with each other via the classic islet hormones – insulin, glucagon and somatostatin [16–19]; via other products secreted by the endocrine cells, such as neurotransmitters or adenine nucleotides and divalent cations that are co-released with insulin [20–23]; and via other less well-known mechanisms, including the generation of gaseous signals such as nitric oxide and carbon monoxide [24–27]. The wide range of intra-islet interactions presumably reflects the requirement for fine-tuning and coordinating secretory responses of many individual islet cells to generate the rate and pattern of hormone secretion appropriate to the prevailing physiologic conditions.
Figure 6.2 Intra-islet autocrine–paracrine interactions. The heterogeneous nature and complex anatomy of the islet enables numerous interactions between islet cells that are mediated by the release of biologically active molecules.
Insulin biosynthesis and storage
The ability to release insulin rapidly in response to metabolic demand, coupled with the relatively slow process of producing polypeptide hormones, means that β-cells are highly specialized for the production and storage of insulin, to the extent that insulin comprises approximately 10% (∼10pg/cell) of the total β-cell protein.
Biosynthesis of insulin
In humans, the gene encoding preproinsulin, the precursor of insulin, is located on the short arm of chromosome 11 [28]. It is 1355 base pairs in length and its coding region consists of three exons: the first encodes the signal peptide at the N-terminus of preproinsulin, the second the B chain and part of the C (connecting) peptide, and the third the rest of the C peptide and the A chain (Figure 6.3). Transcription and splicing to remove the sequences encoded by the introns yields a messenger RNA of 600 nucleotides, translation of which gives rise to preproinsulin, an 11.5-kDa polypeptide. The cellular processes and approximate timescales involved in insulin biosynthesis, processing and storage are summarized in Figure 6.4.
Figure 6.3 Structure of the human insulin gene. The coding region of the human insulin (INS) gene comprises three exons, which encode the signal peptide (SP), the B chain, C peptide and A chain. The exons are separated by two introns (In1 and In2). Beyond the 5’ untranslated region (5’ UT), upstream of the coding sequence, lies a hypervariable region in which three alleles (classes I, II and III) can be distinguished by their size.
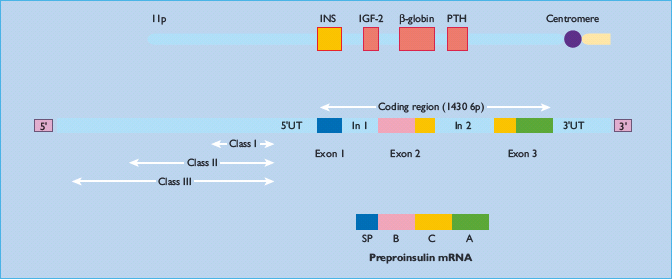
Figure 6.4 The intracellular pathways of (pro)insulin biosynthesis, processing and storage. The molecular folding of the proinsulin molecule, its conversion to insulin and the subsequent arrangement of the insulin hexamers into a regular pattern are shown at the left. The time course of the various processes, and the organelles involved are also shown.
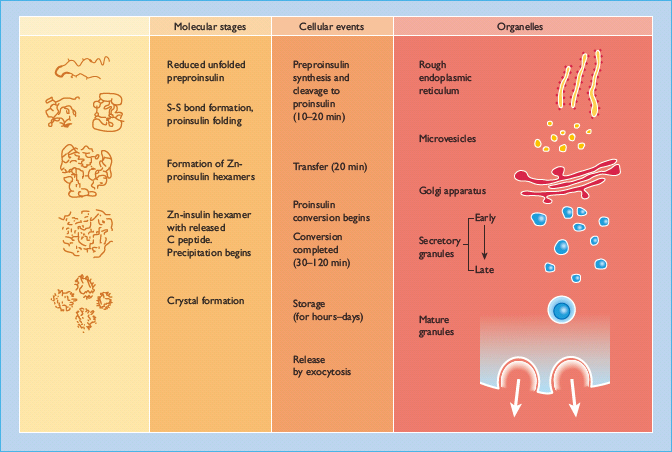
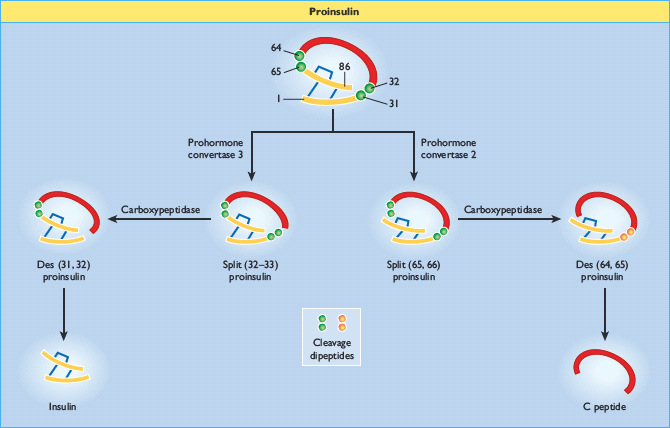
Preproinsulin is rapidly (<1 min) discharged into the cisternal space of the rough endoplasmic reticulum, where proteolytic enzymes immediately cleave the signal peptide, generating proinsulin. Proinsulin is a 9-kDa peptide, containing the A and B chains of insulin (21 and 30 amino acid residues, respectively) joined by the C peptide (30–35 amino acids). The structural conformations of proinsulin and insulin are very similar, and a major function of the C peptide is to align the disulfide bridges that link the A and B chains so that the molecule is correctly folded for cleavage (Figure 6.5). Proinsulin is transported in microvesicles to the Golgi apparatus, where it is packaged into membrane-bound vesicles known as secretory granules. The conversion of proinsulin to insulin is initiated in the Golgi complex and continues within the maturing secretory granule through the sequential action of two endopeptidases (prohormone convertases 2 and 3) and carboxypeptidase H [29], which remove the C peptide chain, liberating two cleavage dipeptides and finally yielding insulin (Figure 6.5). Insulin and C peptide are stored together in the secretory granules and are ultimately released in equimolar amounts by a process of regulated exocytosis. Under normal conditions, >95% of the secreted product is insulin (and C peptide) and <5% is released as proinsulin. However, the secretion of incompletely processed insulin precursors (proinsulin and its “split” products; Figure 6.5) is increased in some patients with type 2 diabetes.
Figure 6.5 Insulin biosynthesis and processing. Proinsulin is cleaved on the C-terminal side of two dipeptides, namely Arg31–Arg32 (by prohormone convertase 3) and Lys64–Arg65 (prohormone convertase 2). The cleavage dipeptides are liberated, so yielding the “split” proinsulin products and ultimately insulin and C peptide.
The β-cell responds to increases in the circulating concentrations of nutrients by increasing insulin production in addition to increasing insulin secretion, thus maintaining insulin stores [30]. Acute (<2 hours) increases in the extracellular concentration of glucose and other nutrients result in a rapid and dramatic increase in the transcription of preproinsulin mRNA and in the rate of proinsulin synthesis [31]. There is a sigmoidal relationship between glucose concentrations and biosynthetic activity, with a threshold glucose level of 2–4 mmol/L. This is slightly lower than the threshold for the stimulation of insulin secretion (∼5 mmol/L), which ensures an adequate reserve of insulin within the β-cell.
Storage and release of insulin
The insulin secretory granule has a typical appearance in electron micrographs, with a wide space between the crystalline electron-opaque core and its limiting membrane (Figure 6.1c). The major protein constituents of the granules are insulin and C peptide, which account for approximately 80% of granule protein [31], with numerous minor components including peptidases, peptide hormones and a variety of (potentially) biologically active peptides of uncertain function [32,33]. Insulin secretory granules also contain high concentrations of divalent cations, such as zinc (∼20 mmol/L), which is important in the crystallization and stabilization of insulin within the granule [34,35]. The intra-granular function(s) of calcium (∼120mmol/L) and magnesium (∼70 mmol/L) are uncertain, but as they are co-released with insulin on exocytosis of the secretory granule contents they may have extracellular signaling roles via the cell surface calcium-sensing receptor [20]. Similarly, the adenine nucleotides found in insulin secretory granules (∼10 mmol/L) may have a signaling role when they are released into the extracellular space [21].
The generation of physiologically appropriate insulin secretory responses requires complex mechanisms for moving secretory granules from their storage sites within the cell to the specialized sites for exocytosis on the inner surface of the plasma membrane, and the role of cytoskeletal elements, notably microtubules and microfilaments, in the intracellular translocation of insulin storage granules has been studied extensively [36,37]. Microtubules are formed by the polymerization of tubulin subunits and normally form a network radiating outwards from the perinuclear region [38]. The microtubular network is in a process of continual remodeling and the dynamic turnover of tubulin, rather than the total number of microtubules, is important for the mechanism of secretion. The microtubule framework may provide the pathway for the secretory granules but microtubules do not provide the motive force [39] so other contractile proteins are likely to be involved. Actin is the constituent protein of microfilaments which exists in cells as a globular form of 43 kDa and as a filamentous form which associates to form microfilaments. Microfilament polymerization is regulated by agents that alter rates of insulin secretion [40], and the pharmacologic disruption of microfilament formation inhibits insulin secretion. Myosin light and heavy chains are expressed at high concentrations in β-cells, suggesting that actin and myosin may interact to propel granules along the microtubular network. It also seems likely that other molecular motors, including kinesin and dynein [41–43], are also involved in the movement of secretory granules, and perhaps other organelles, in β-cells.
Insulin is released from secretory granules by exocytosis, a process in which the granule membrane and plasma membrane fuse together, releasing the granule contents into the interstitial space. Much of our knowledge of the molecular mechanisms of exocytosis is derived from studies of neurotransmitter release from nerve cells, and similar mechanisms operate in β-cells. The docking of the granules at the inner surface of the plasma membrane is via the formation of a multimeric complex of proteins known as the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex, which consists of proteins associated with secretory granules and the plasma membrane, and soluble fusion proteins [44,45]. The docked granules will only fuse with the membrane and release their contents in the presence of elevated levels of intracellular calcium which is sensed by synaptotagmins, a class of calcium-binding granule proteins [44,46]. Secretory granules are distributed throughout the β-cell cytoplasm (Figure 6.1c), and it is likely that the transport of granules from distant sites to the plasma membrane is regulated independently from the final secretory process, with a reservoir of granules stored close to the inner surface of the plasma membrane. Fusion of this “readily-releasable” pool of granules may account for the rapid first-phase release of insulin in response to glucose stimulation [39,40]. Direct electrophysiologic measurements have demonstrated that the β-cell exocytotic response consists of a short-lived first phase, with a very rapid rate of granule exocytosis, followed by a sustained second phase with a slower rate of exocytosis [45,46].
Regulation of insulin secretion
To ensure that circulating levels of insulin are appropriate for the prevailing metabolic status β-cells are equipped with mechanisms to detect changes in circulating nutrients, in hormone levels, and in the activity of the autonomic nervous system. Moreover, β-cells have fail-safe mechanisms for coordinating this afferent information and responding with an appropriate secretion of insulin. The major physiologic determinant of insulin secretion in humans is the circulating concentration of glucose and other nutrients, including amino acids and fatty acids. These nutrients possess the ability to initiate an insulin secretory response, so when nutrients are being absorbed from the gastrointestinal system the β-cell detects the changes in circulating nutrients and releases insulin to enable the uptake and metabolism or storage of the nutrients by the target tissues. The consequent decrease in circulating nutrients is detected by the β-cells, which switch off insulin secretion to prevent hypoglycemia. The responses of β-cells to nutrient initiators of insulin secretion can be modified by a variety of hormones and neurotransmitters which act to amplify, or occasionally inhibit, the nutrient-induced responses (Table 6.1). Under normoglycemic conditions these agents have little or no effect on insulin secretion, a mechanism that prevents inappropriate secretion of insulin which would result in potentially harmful hypoglycemia. These agents are often referred to as potentiators of insulin secretion to distinguish them from nutrients which initiate the secretory response. The overall insulin output depends on the relative input from initiators and potentiators at the level of individual β-cells, on the synchronization of secretory activity between β-cells in individual islets, and on the coordination of secretion between the hundreds of thousands of islets in a human pancreas. This section considers the mechanisms employed by β-cells to recognize and respond to nutrient initiators and non-nutrient potentiators of insulin secretion.
Table 6.1 Non-nutrient regulators of insulin secretion.
| Stimulators | Inhibitors |
| Islet products | |
| Glucagon | SST-14 |
| Adenine nucleotides | Ghrelin |
| Divalent cations | |
| Neurotransmitters | |
| Acetylcholine | Norepinephrine |
| VIP | Dopamine |
| PACAP | NPY |
| GRP | Galanin |
| Gastrointestinal hormones | |
| CCK | SST-28 |
| GIP | Ghrelin |
| GLP-1 | |
| Adipokines | |
| Adiponectin | Leptin |
| Resistin |
CCK, cholecystokinin; GIP, glucose-dependent insulinotropic peptide; GLP-1, glucagon-like peptide-1; GRP, gastrin-releasing polypeptide; NPY, neuropeptide Y; PACAP, pituitary adenylate cyclase activating polypeptide; SST; somatostatin; VIP, vasoactive intestinal polypeptide.
Nutrient-induced insulin secretion
Nutrient metabolism
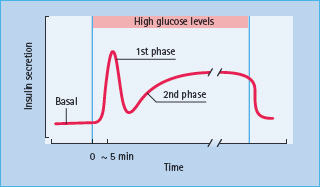
Pancreatic β-cells respond to small changes in extracellular glucose concentrations within a narrow physiologic range and the mechanisms through which β-cells couple changes in nutrient metabolism to regulated exocytosis of insulin are becoming increasingly well understood. Glucose is transported into β-cells via high capacity glucose transporters (GLUT; GLUT2 in rodents, GLUT1, 2 and 3 in humans [47,48]), enabling rapid equilibration of extracellular and intracellular glucose concentrations. Once inside the β-cell glucose is phosphorylated by glucokinase which acts as the “glucose sensor,” coupling insulin secretion to the prevailing glucose level [49]. The dose–response curve of glucose-induced insulin secretion from isolated islets is sigmoidal (Figure 6.6) and is determined primarily by the activity of glucokinase. Concentrations of glucose below 5 mmol/L do not affect rates of insulin release, and the rate of secretion increases progressively at extracellular glucose levels between 5 and ∼15 mmol/L, with halfmaximal stimulation at ∼8 mmol/L. The time-course of the insulin secretory response to elevated glucose is characterized by a rapidly rising but transient first phase, followed by a maintained and prolonged second phase, as shown in Figure 6.7. This profile of insulin secretion is obtained whether insulin levels are measured following a glucose load in vivo, or whether the secretory output from the perfused pancreas or isolated islets is assessed, suggesting that the characteristic biphasic secretion pattern is an intrinsic property of the islets.
Figure 6.6 Glucose-induced insulin secretion from islets of Langerhans. No stimulation is seen below a threshold value of approximately 5mmol/L glucose. Potentiators amplify insulin secretion at stimulatory concentrations of glucose, but are ineffective at subthreshold glucose levels.
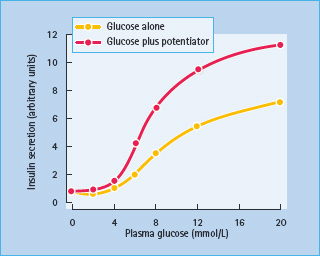
Figure 6.7 Glucose-induced insulin release in vitro. The figure shows the pattern of glucose-induced insulin secretion from perfused pancreas, in response to an increase in the glucose concentration. An acute first phase, lasting a few minutes, is followed by a sustained second phase of secretion which persists for the duration of the high-glucose stimulus.
ATP-sensitive potassium channels and membrane depolarization
In the absence of extracellular glucose, the β-cell membrane potential is maintained close to the potassium equilibrium potential by the efflux of potassium ions through inwardly rectifying potassium channels. These channels were called ATP-sensitive potassium (KATP) channels because application of ATP to the cytosolic surface of β-cell membrane patches resulted in rapid, reversible inhibition of resting membrane permeability to potassium ions [50]. This property of the KATP channel is pivotal in linking glucose metabolism to insulin secretion. Thus, it is now well established that ATP generation following glucose metabolism, in conjunction with concomitant lowering of ADP levels, leads to closure of β-cell KATP channels. Channel closure and subsequent reduction in potassium efflux promotes depolarization of the β-cell membrane and influx of calcium ions through voltage-dependent L-type calcium channels. The resultant increase in cytosolic Ca2+ triggers the exocytosis of insulin secretory granules, thus initiating the insulin secretory response (Figure 6.8).
Figure 6.8 Intracellular mechanisms through which glucose stimulates insulin secretion. Glucose is metabolized within the β-cell to generate ATP, which closes ATP-sensitive potassium channels in the cell membrane. This prevents potassium ions from leaving the cell, causing membrane depolarization, which in turn opens voltage – gated calcium channels in the membrane and allows calcium ions to enter the cell. The increase in cytosolic calcium initiates granule exocytosis. Sulfonylureas act downstream of glucose metabolism, by binding to the SUR1 component of the KATP channel (inset). GLUT, glucose transporter.
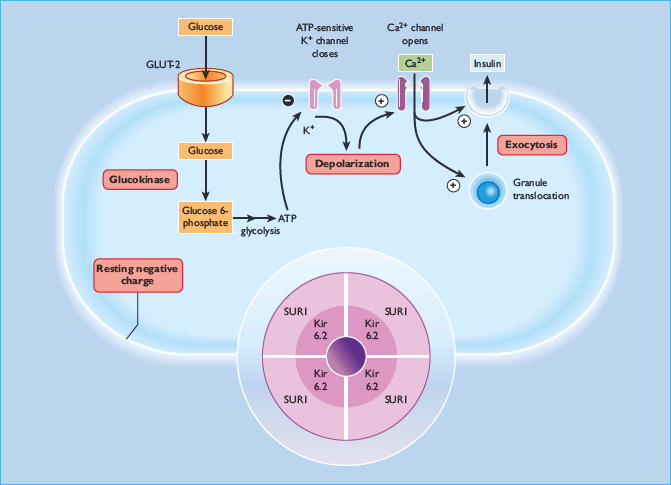
At around the time that the KATP channels were established as the link between the metabolic and electrophysiologic effects of glucose, they were also identified as the cellular target for sulfonylureas. The capacity of sulfonylureas to close KATP channels explains their effectiveness in type 2 diabetes where the β-cells no longer respond adequately to glucose, as the usual pathway for coupling glucose metabolism to insulin secretion is bypassed. The β-cell KATP channel is a hetero-octamer formed from four potassium channel subunits (termed Kir6.2) and four sulfonylurea receptor subunits (SUR1) [51]. The Kir6.2 subunits form the pore through which potassium ions flow and these are surrounded by the SUR1 subunits which have a regulatory role (Figure 6.8, inset). ATP and sulfonylureas induce channel closure by binding to Kir6.2 and SUR1 subunits, respectively, while ADP activates the channels by binding to a nucleotide-binding domain on the SUR1 subunit. Diazoxide, an inhibitor of insulin secretion, also binds to the SUR1 subunit to open the channels. The central role of KATP channels in β-cell glucose recognition makes them obvious candidates for β-cell dysfunction in type 2 diabetes. Earlier studies in patients with type 2 diabetes, maturity-onset diabetes of the young (MODY) or gestational diabetes, failed to detect any Kir6.2 gene mutations that compromised channel function [52,53]. Since then, larger scale studies of variants in genes encoding Kir6.2 and SUR1 have demonstrated polymorphisms associated with increased risk of type 2 diabetes [54]. Similarly, activating mutations in the Kir6.2 gene have recently been shown to be causal for cases of permanent neonatal diabetes (PNDM) [55], which has enabled insulin-dependent PNDM patients to achieve glycemic control with sulfonylurea treatment alone. In contrast, loss of β-cell functional KATP channel activity has been implicated in the pathogenesis of persistent hyperinsulinemic hypoglycemia of infancy (PHHI) [56], a condition characterized by hypersecretion of insulin despite profound hypoglycemia. Numerous mutations in both the Kir6.2 and SUR1 subunits have been identified in patients with PHHI and these are thought to be responsible for the severe impairment in glucose homeostasis in these individuals [57].
Calcium and other intracellular effectors
Intracellular calcium is a principal effector of the nutrient-induced insulin secretory response, linking depolarization with exocytosis of insulin secretory granules (Figure 6.8). A large electrochemical concentration gradient (∼10 000-fold) of calcium is maintained across the β-cell plasma membrane by a combination of membrane-associated calcium extruding systems and active calcium sequestration within intracellular organelles. The major route through which calcium is elevated in β-cells is by influx of extracellular calcium through voltage-dependent L-type calcium channels that open in response to β-cell depolarization, and it has been estimated that each β-cell contains about 500 L-type channels [58].
Studies with permeabilized β-cells have demonstrated that elevations in intracellular calcium are alone sufficient to initiate insulin secretion [59] and conditions that elevate intracellular calcium usually stimulate insulin release. An increase in cytosolic calcium is essential for the initiation of insulin secretion by glucose and other nutrients: preventing calcium influx by removal of extracellular calcium or by pharmacologic blockade of voltage-dependent calcium channels abolishes nutrient-induced insulin secretion. Glucose and other nutrients also induce a calcium-dependent activation of β-cell phospholipase C (PLC) [60], leading to the generation of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG), both of which serve second-messenger functions in β-cells [61]. The generation of IP3 leads to the rapid mobilization of intracellular calcium, but the significance of this in secretory responses to nutrients is uncertain, and it is likely to have little more than a modulatory role, amplifying the elevations in cytosolic calcium concentration induced by the influx of extracellular calcium.
The elevations in intracellular calcium are transduced into the regulated secretion of insulin by intracellular calcium sensing systems within β-cells. Important among these are the calcium-dependent protein kinases, which include myosin light chain kinases, the calcium/phospholipid-dependent kinases and the calcium/calmodulin-dependent kinases (CaMKs). CaMKs are protein kinases that are activated in the presence of calcium and the calcium-binding protein, calmodulin, and a number of studies have implicated CaMK II in insulin secretory responses [61]. It has been proposed that CaMK II activation is responsible for the initiation of insulin secretion in response to glucose and other nutrients, and for enhancing nutrient-induced secretion in response to receptor agonists that elevate intracellular calcium [61]. Cytosolic PLA2 (cPLA2) is another β-cell calcium-sensitive enzyme. It is activated by concentrations of calcium that are achieved in stimulated β-cells, and it generates arachidonic acid (AA) by the hydrolysis of membrane phosphatidylcholine. AA is capable of stimulating insulin secretion in a glucose and calcium-independent manner, and it is further metabolized in islets by the cyclo-oxygenase (COX) pathways to produce prostaglandins and thromboxanes, and by the lipoxygenase (LOX) pathways to generate hydroperoxyeicosatetrenoic acids (HPETES), hydroxyeicosatetrenoic acids (HETES) and leukotrienes.
The precise roles(s) of AA derivatives in islet function remains uncertain because experimental investigations have relied on COX and LOX inhibitors of poor specificity, but earlier reports that prostaglandins are largely inhibitory in rodent islets [62] have not been supported by more recent studies using human islets [63]. Calcium sensors are also important at the later stages of the secretory pathway, where the calcium-sensitive synaptotagmin proteins are involved in the formation of the exocytotic SNARE complex, as described above, to confer calcium sensitivity on the initiation and rate of exocytotic release of insulin secretory granules [44].
The elevations in intracellular calcium induced by nutrients activate other effector systems in β-cells, including PLC and cPLA2, as discussed above, and calcium-sensitive adenylate cyclase isoforms, which generate cyclic AMP from ATP. Although these signaling systems are of undoubted importance in the regulation of β-cells by non-nutrients, their role in nutrient-induced insulin secretion is still uncertain. Thus, DAG generated by glucose-induced PLC activation has the potential to activate some protein kinase C (PKC) isoforms. PKC was first identified as a calcium and phospholipid sensitive, DAG-activated protein kinase, but it has become apparent that some isoforms of PKC require neither calcium nor DAG for activation. The isoforms are classified into three groups: calcium and DAG sensitive (conventional); calcium independent, DAG-sensitive (novel); and calcium and DAG independent (atypical), and β-cells contain conventional, atypical and novel PKC isoforms [64]. The early literature on the role of PKC in nutrient-induced insulin secretion is confusing but several studies have shown that glucose-induced insulin secretion is maintained under conditions where DAG-sensitive PKC isoforms are depleted, suggesting that conventional and novel PKC isoforms are not required for insulin secretion in response to glucose [61,65].
The role of cyclic AMP in the insulin secretory response to nutrients is similarly unclear. Cyclic AMP has the potential to influence insulin secretion by the activation of cyclic AMP-dependent protein kinase A (PKA), or via the cyclic AMP-regulated guanine nucleotide exchange factors known as exchange proteins activated by cyclic AMP (EPACs) [66]. However, elevations in β-cell cyclic AMP do not stimulate insulin secretion at substimulatory glucose concentrations, and the secretagogue effects of glucose can be maintained in the presence of competitive antagonists of cyclic AMP binding to PKA or EPACs [67]. These observations suggest that cyclic AMP does not act as a primary trigger of nutrient-stimulated β-cell secretory function, but more recent observations linking glucose-induced oscillations in β-cell cyclic AMP to oscillations in insulin secretion [68] suggest that a role for this messenger system in nutrient-induced insulin secretion cannot be ruled out.
KATP channel-independent pathways
Since the early reports linking KATP channel closure to the exocytotic release of insulin, it has become apparent that β-cells also possess a KATP channel-independent stimulus–secretion coupling pathway: this is termed the amplifying pathway to distinguish it from the triggering pathway that is activated by KATP channel closure [69]. Studies in which β-cell calcium is elevated by depolarization and KATP channels maintained in the open state by diazoxide have indicated that glucose, at concentrations as low as 1–6 mmol/L, is still capable of stimulating insulin secretion [70]. The mechanisms by which glucose stimulates insulin secretion in a KATP channel-mdependent manner have not been established [71]. However, it is clear that glucose must be metabolized and there is convincing evidence that changes in adenine nucleotides are involved [72], while it has been established that activation of PKA and PKC is not required. It has been suggested that the KATP-independent amplifying pathway is impaired in type 2 diabetes and that identification of novel therapeutic strategies targeted at this pathway may be beneficial in restoring β-cell function in patients with type 2 diabetes [69].
Amino acids
Several amino acids stimulate insulin secretion in vivo and in vitro. Most require glucose, but some, such as leucine, lysine and arginine, can stimulate insulin secretion in the absence of glucose, and therefore qualify as initiators of secretion. Leucine enters islets by a sodium-independent transport system and stimulates a biphasic increase in insulin release. The effects of leucine on β-cell membrane potential, ion fluxes and insulin secretion are similar to, but smaller than, those of glucose [73]. Thus, metabolism of leucine within β-cells decreases the potassium permeability, causing depolarization and activation of L-type calcium channels through which calcium enters the β-cells and initiates insulin secretion. Leucine is also able to activate the amplifying pathway of insulin secretion in a KATP channel-independent manner, as described above for glucose. The charged amino acids, lysine and arginine, cross the β-cell plasma membrane via a transport system specific for cationic amino acids. It is generally believed that the accumulation of these positively charged molecules directly depolarizes the β-cell membrane leading to calcium influx.
Regulation of insulin secretion by non-nutrients
The complex mechanisms that have evolved to enable changes in extracellular nutrients to initiate an exocytotic secretory response are confined to pancreatic β-cells, and perhaps to a subset of hypothalamic neurons [74]. However, the mechanisms that β-cells use to recognize and respond to non-nutrient potentiators of secretion are ubiquitous in mammalian cells, and so are covered only briefly in this section, followed by a review of the physiologically relevant non-nutrient regulators of β-cell function.
Most, if not all, non-nutrient modulators of insulin secretion influence the β-cell by binding to and activating specific receptors on the extracellular surface. Because of its central role in coordinating whole body fuel homeostasis the β-cell expresses receptors for a wide range of biologically active peptides, glycoproteins and neurotransmitters (Table 6.1). However, receptor occupancy generally results in the activation of a limited number of intracellular effector systems, which were introduced in the section on nutrient-induced insulin section (Figures 6.9 and 6.10).
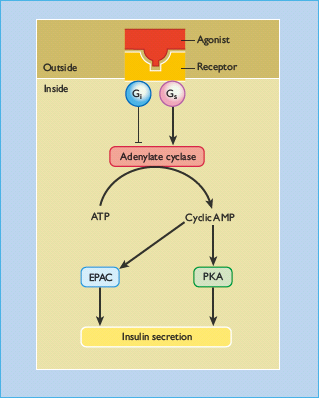
Figure 6.9 Adenylate cyclase and the regulation of insulin secretion. Some receptor agonists (e.g. glucagon, glucagon – like peptide 1, pituitary adenylate cyclase activating polypeptide) bind to cell-surface receptors that are coupled to adenylate cyclase (AC) via the heterotrimeric GTP-binding protein Gs. Adenylate cyclase hydrolyses ATP to generate adenosine 5’ cyclic monophosphate (cyclic AMP), which activates protein kinase A (PKA) and exchange proteins activated by cyclic AMP (EPACs). Both of these pathways potentiate glucose-stimulated insulin secretion. Glucose also activates adenylate cyclase, but increases in intracellular cyclic AMP levels in response to glucose are generally smaller than those obtained with receptor agonists. Some inhibitory agonists (e.g. norepinephrine, somatostatin) bind to receptors that are coupled to adenylate cyclase via the inhibitory GTP-binding protein Gi, resulting in reduced adenylate cyclase activity and a decrease in intracellular cyclic AMP.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


