Chapter 10 Investigation of megaloblastic anaemia
cobalamin, folate and metabolite status
Cobalamin absorption and metabolism
Cobalamin in the human diet is a bacterial product ingested and stored by animals and strict vegans are therefore liable to deficiency. The prevalence of cobalamin deficiency, as defined by serum vitamin B12 <200 ng/l and methylmalonic acid >0.27 μmol/l, is 1.6% of subjects over 51 years of age in the 2001–2004 National Health and Nutrition Examination Survey1 in the USA. Of this age group, 3.2% have a low serum B12 <200 ng/l. Ingested cobalamin is released from food proteins by pepsin and acid and bound initially by transcobalamin I or haptocorrin (R binder). This binder also binds other cobinamides in the diet which are metabolically inert. Pancreatic enzymes release the cobalamin from transcobalamin I and permit binding by intrinsic factor. The intrinsic factor–cobalamin complex is attached to cubam, a multiligand receptor, which is a combination of cubilin2 and amnionless and is taken up by endocytosis, into the ileal cell. The cobalamin is then released from the endosome and bound to transcobalamin II (holotranscobalamin) in the ileal cell and exported into the portal circulation. Cobalamin undergoes enterohepatic circulation via the liver and bile ducts with 1.4 μg/day excreted in the bile, of which 1 μg/day is reabsorbed in the ileum. Holotranscobalamin is the active form of cobalamin and is taken up by holotranscobalamin receptors (TCII receptors) on cells throughout the body, particularly liver, kidney and bone marrow cells. At the cellular level in the target tissue the holotranscobalamin undergoes endocytosis via the transmembrane TCII receptor. Holotranscobalamin then undergoes lysosomal degradation, releasing cobalamin for metabolic reactions.
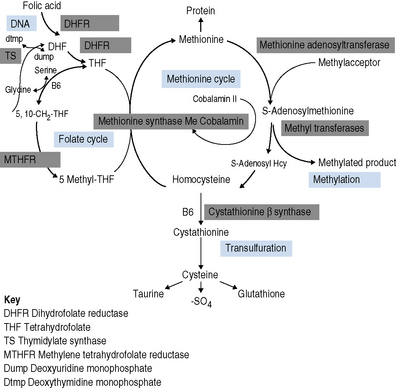
Cobalamin is a cofactor in two important biochemical reactions. In the first, methylcobalamin acts as a cofactor for methionine synthase in the production of methionine from homocysteine. The remethylation of cobalamin requires the donation of the methyl group from methyltetrahydrofolate as it is converted to tetrahydrofolate, thus linking cobalamin to folate and 1-carbon metabolism. The second cobalamin reaction occurs in the mitochondrion. Cobalamin is converted to adenosylcobalamin, a cofactor for the enzyme methylmalonyl-CoA mutase, which converts methylmalonyl-CoA (the product of propionate metabolism) to succinyl-CoA. Methionine produced in the first reaction is converted to adenosylmethionine and is a vital source of methyl groups critical for a series of methylation reactions involving proteins, phospholipids, neurotransmitters, RNA and DNA. In cobalamin deficiency, methylmalonic acid and homocysteine levels are therefore elevated. Reduced methionine synthesis is thought to result in a decrease in methylation of myelin basic protein, resulting in the neuropathies associated with cobalamin deficiency which are irreversible once subacute combined degeneration3 of the cord has occurred.
Folate absorption and metabolism
There is sufficient retention of folate by the renal tubules to prevent urinary vitamin loss; this is achieved by megalin uptake of filtered folate-binding protein4 and bound folate. Cubam,2 which binds intrinsic factor-cobalamin complex, is also important in the uptake of albumen from the renal tubules, which may also contribute to folate retention.
Folates participate in 1-C metabolism and thereby facilitate the essential cellular metabolism of methionine, serine, glycine, choline and histidine in the biosynthesis of purine and deoxythymidine monophosphate (dTMP) in the synthesis of pyrimidines and thus DNA (Fig. 10.1).
Rationale for investigation of cobalamin or folate status
Investigation of the vitamin B12 and folate status of individuals is not restricted to investigation of individuals with classical features of megaloblastic anaemia alone because neuropathy and neuro-psychiatric changes may occur in B12 deficiency in the absence of macrocytosis or anaemia.5–9 The finding that folate supplementation reduced the incidence of neural tube defects10 by 25–46% in the USA and Canada highlights the importance of defining optimum population folate levels. Increased plasma homocysteine and serum methylmalonic acid (MMA) levels have been advocated11–14 as sensitive indicators of folate and cobalamin deficiency that may be subclinical. Introduction of metabolite testing to routine laboratory practice was limited in the past as a result of technical difficulty in measurement but is now becoming more available, though the clinical benefit of this approach has been questioned by some authors.15,16 Elevated homocysteine levels are an independent vascular disease risk factor17 and are also associated with risk of idiopathic venous thrombosis.18 In the USA, dietary supplementation with folate was introduced in 1998 to reduce neural tube defects and by lowering homocysteine levels may achieve a further health gain in reduced myocardial infarction and stroke. Food supplementation with folate remains a controversial step in European countries because of a possible increased risk of cancers19,20 (see Scientific Advisory Committee on Nutrition, at: www.sacn.gov.uk, for update). Increased plasma homocysteine levels occur in both B12 and folate deficiency as a result of reduction in methionine synthesis (Fig. 10.1). Some laboratories have utilized homocysteine as an initial screening test for abnormalities of cobalamin and folate metabolism which may be particularly appropriate for investigation of suspected inherited cobalamin or folate disorders in children.21,22 MMA measurement though not widely available in the UK, requiring gas chromatography-mass spectrometry (GC-MS), is available at a limited number of university departments. In contrast, plasma homocysteine measurement by high-performance liquid chromatography (HPLC) and commercial enzyme immunoassays is widely available. The limitations of total serum B12 measurement have been highlighted by studies that showed poor positive predictive value (i.e. healthy persons with a low level or low cobalamin levels with no evidence of deficiency) and <100% negative predictive value of 95% (i.e. 5% clinically deficient with normal level).15,23 In addition, some publications have highlighted the presence of severe cobalamin deficiency concurrent with a normal cobalamin level using some of the current commercial assays.24,25 The introduction of holotranscobalamin assays,26–28 now available on an Abbott automated immunoassay platform, provides a readily accessible method of assessing the physiologically active form of cobalamin rather than the less relevant total B12. The advent of a commercial holotranscobalamin assay occurred over 20 years after depletion of serum holotranscobalamin II was identified as an early sign of cobalamin deficiency.29 Adoption of new methodology in laboratory medicine in the UK requires careful evaluation in a clinical setting and this research appears inhibited by lack of training in research methodology and financial constraints in service laboratories. Serum methylmalonic acid is increased in renal impairment and 7.9% of subjects over 51 have levels in excess of 0.27 μmol/l.1 MMA must therefore be used in conjunction with serum cobalamin and holotranscobalamin and must be interpreted in the light of renal function. Urinary MMA measurement can be used to compensate for the impact of renal impairment.
Haematological features of megaloblastic anaemia
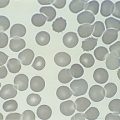
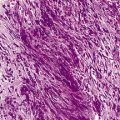
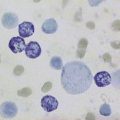
Megaloblastic anaemia resulting from impaired DNA synthesis is characterized by the presence of megaloblastic red cell precursors in the bone marrow and occasionally also in the blood. Megaloblasts have a characteristic chromatin pattern (Fig. 10.2) and increased cytoplasm as a result of asynchrony of nuclear and cytoplasmic maturation with a relatively immature nucleus for the degree of cytoplasmic haemoglobinization. The delay in nuclear maturation caused by delay in DNA synthesis resulting from lack of vitamin B12 or folate is also seen in all lineages, particularly granulocytic marrow precursors with giant metamyelocytes (Fig. 10.3) and hyperlobated neutrophils with increased lobe size as well as number of nuclear segments (see Chapter 5, Fig. 5.10). In severe pernicious anaemia, a progressive increase in mean red cell volume (MCV) up to 130 fl occurs, with oval macrocytes, poikilocytes and hypersegmentation of neutrophils (>5% with more than five nuclear lobes).31 The neutrophil hypersegmentation index is an equivalent automated parameter on some cell counters, although hypersegmentation does not always respond to a therapeutic trial. The mean platelet volume is decreased and there is increased platelet anisocytosis, as detected by the platelet distribution width (PDW). The MCV falls to 110–120 fl as megaloblastic change advances. Howell–Jolly bodies and basophilic stippling are seen in the red cells.
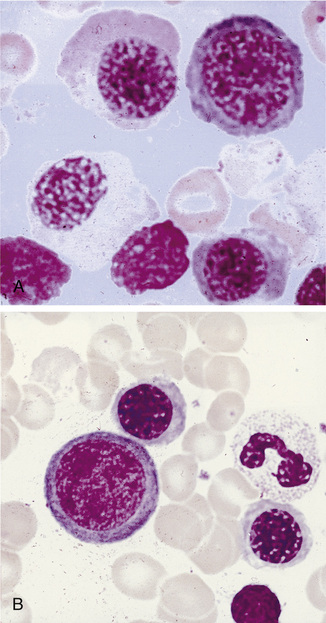
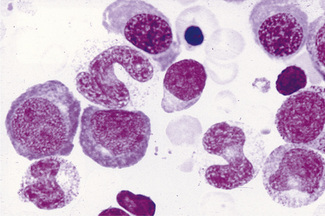
Figure 10.3 Photomicrograph of bone marrow film stained by May–Grünwald–Giemsa showing giant metamyelocytes.
Differential Diagnosis of Macrocytic Anaemia
Macrocytic red cells are also seen in myelodysplastic syndromes, which can be suspected from the presence of hypogranular neutrophils (see Chapter 5, Fig. 5.76) or monocytosis. Excess alcohol consumption results in an increased MCV as a result of round macrocytes, although rarely does it go higher than 110 fl unless coexisting folate deficiency is present. Hypothyroidism, liver disease, aplastic anaemia, rare inherited orotic aciduria or Lesch–Nyhan syndrome also have a high MCV. Automated reticulocyte counts facilitate detection of increased red cell turnover and high MCV as a result of haemolysis or bleeding. Coexisting iron deficiency or thalassaemia trait may mask macrocytic changes, although a high red cell distribution width indicates anisocytosis and the need for blood film review. Congenital dyserythropoietic anaemias types I and III and erythroleukaemia exhibit some features of megaloblastic erythropoiesis that are unrelated to B12 and folate. Drugs interfering with DNA synthesis (e.g. azathioprine, zidovudine or hydroxycarbamide) result in macrocytosis and megaloblastic erythropoiesis. Anticonvulsant therapy interferes with folate metabolism,32 whereas the impact of oral contraceptives on folate absorption and metabolism is controversial.33 Prolonged nitrous oxide anaesthesia destroys methylcobalamin and causes acute megaloblastic change.34 Methotrexate inhibits dihydrofolate reductase and toxicity can be reversed with folinic acid, which is already in the tetrahydrofolate form and effectively reverses the metabolic block, which is not achieved solely with folic acid.
Testing strategy for suspected cobalamin or folate deficiency
Microbiological cobalamin and folate assays and competitive radiodilution binding assays for measurement of cobalamin and folate, which were often performed together, have largely been replaced by separate analysis by automated binding assays. The application of a suitable testing strategy for patients suspected of having cobalamin or folate deficiency is shown in Tables 10.1–10.3.
Table 10.1 Significance of clinical details
| Symptoms or signs | Possible significance | |
|---|---|---|
| Tiredness, palpitations, pallor | Anaemia | |
| Slight jaundice | Ineffective erythropoiesis | |
| Neurological | ||
| Cognitive impairment, optic atrophy, loss of vibration sense, joint position sense; plantar responses normal or abnormal; tendon reflexes depressed or increased | Cobalamin deficiency, subacute combined degeneration of the spinal cord and sensori/motor peripheral neuropathies | |
| Dietary and gastrointestinal history | ||
| Vegetarian or vegan; poor nutrition (e.g. tea and toast diet in elderly or students); dietary fads | Low iron stores and iron deficiency | |
| Cobalamin deficiency in babies born to mothers who are vegans | ||
| Folate deficiency (often with iron deficiency) | ||
| Weight loss, bloating and steatorrhoea, particularly nocturnal bowel movements | Features of malabsorption and folate deficiency, e.g. due to coeliac disease, tropical sprue | |
| Mouth ulcers, abdominal pain, perianal ulcers, fistulae | Terminal ileal Crohn’s disease – cobalamin deficiency | |
| Glossitis, angular cheilosis and koilonychia | Cobalamin and combined iron deficiency | |
| Alcohol history | Poor diet and interference with folate metabolism | |
| History of autoimmune disease in patient or family | ||
| Hypothyroidism, pernicious anaemia or coeliac disease | Increased likelihood of pernicious anaemia or coeliac disease | |
| Surgery | ||
| Gastrectomy/bowel resection | Cobalamin deficiency usually 2 years post-gastrectomy | |
| Ileal disease resulting in cobalamin deficiency | ||
| Blind loop syndromes | ||
| Physical appearance | ||
| Grey hair, blue eyes, vitiligo | Association with pernicious anaemia | |
| Pregnancy | Increased iron and folate requirements. | |
| Cobalamin levels fall by 30% in the 3rd trimester | ||
| Holotranscobalamin levels are unaltered in late pregnancy | ||
| Malabsorptive syndrome | ||
| Tropical sprue, bacterial overgrowth, fish tape worm in Scandinavian countries | Combined folate and iron deficiency | |
| Cobalamin deficiency | ||
| Drug history | See text | |
| Other haematological disorders | ||
| Myeloproliferative neoplasms, haemolytic anaemias, leukaemias | Increased folate utilization may result in folate deficiency | |
| Myeloma | Paraprotein interference with cobalamin assays resulting in falsely low cobalamin levels, which normalize on treatment of myeloma | |
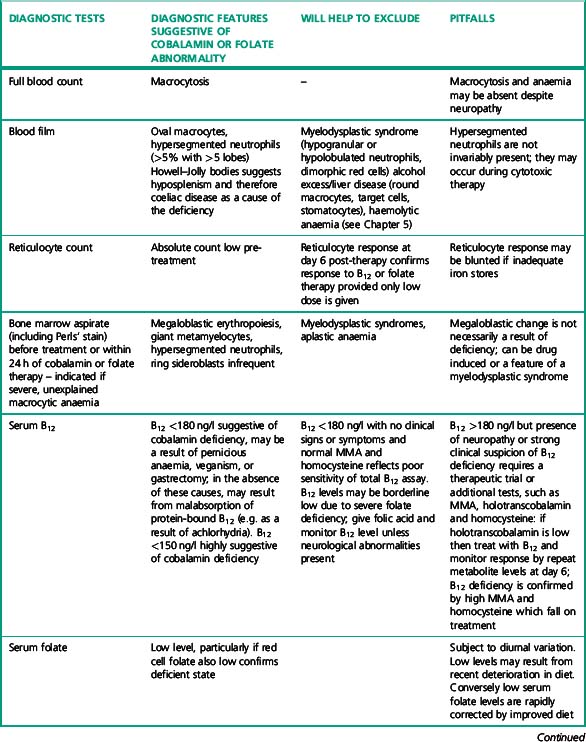
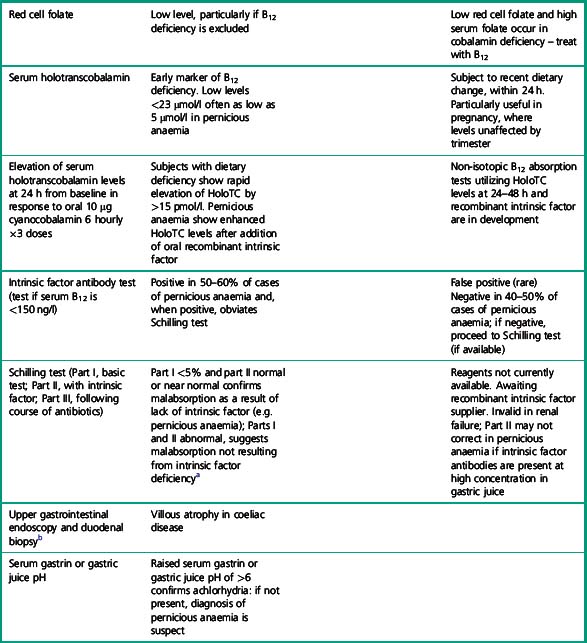

Table 10.3 Clinical and laboratory checklist for diagnosis of pernicious anaemia
| Laboratory criteria | Clinical criteria | |
|---|---|---|
| Minor criteria | Macrocytosis | Parasthesiae, numbness or ataxia |
| Anaemia | Hypothyroidism | |
| Raised plasma homocysteine | Vitiligo | |
| Gastric pH above 6 Raised serum gastrin | Family history of pernicious anaemia or hypothyroidism | |
| Positive gastric parietal cell antibody | ||
| Major criteria | Low serum B12 (<180 ng/l) or raised serum methylmalonic acid (>0.75 μmol/l) in presence of normal renal function | |
| Megaloblastic anaemia not resulting from folate deficiency | ||
| Positive intrinsic factor antibodies using high-specificity test. | ||
| Holotranscobalamin level <23 μmol/l | ||
| Reference standard criteria | Schilling testa shows malabsorption of oral cyanocobalamin corrected by coadministration of intrinsic factor |
a Reagents for Schilling tests currently unavailable. A non-isotopic B12 absorption test utilizing recombinant intrinsic factor and holotranscobalamin measurement is under development.
Table 10.1 highlights the important clinical details that should be elicited by the clinician and submitted with the request to assist the laboratory in interpretation of the numeric results of cobalamin and folate assays. Ideally, test requests should not be accepted without this information, which could be incorporated into electronic order communication from user to laboratory.
Table 10.2 lists the important laboratory investigations that should be performed – results must not be reported in isolation from other laboratory results and clinical details. If investigations are performed in different laboratories, authorization and release of results requires access to all laboratory data on the individual patient. Laboratory information systems should facilitate this cross-disciplinary access. For example, intrinsic factor antibody results should be available to haematology or clinical chemistry laboratories undertaking cobalamin, MMA or homocysteine assays.
Table 10.3 provides a list of clinical and laboratory features for diagnosis of pernicious anaemia. These criteria avoid undue reliance on a single B12 assay and should help clinicians to make a diagnosis even when some critical tests, e.g. Schilling tests, are not available.
In view of the lack of specificity and sensitivity of serum cobalamin assays and frequent lack of availability of other diagnostic tests, basing the diagnosis of pernicious anaemia, which requires lifelong parenteral B12 therapy, solely on laboratory results, is not straightforward. A checklist of laboratory and clinical diagnostic criteria, as shown in Table 10.2, helps to achieve a greater degree of diagnostic certainty than any single diagnostic test and permits the diagnosis to be made even when a single diagnostic test is anomalous or unavailable. Clinical and other laboratory criteria thus provide additional supportive evidence of an autoimmune aetiology, even if the more demanding diagnostic laboratory criteria are not met.
Limitations of Cobalamin Assays
Sensitivity and Specificity of Cobalamin and Holotranscobalamin Assays
Utility of receiver operator characteristic curves
There has been little data on sensitivity and specificity of current B12 assays, due to the difficulty in defining a truly deficient study population. Some authors have suggested B12 assays and measurement of methylmalonic acid are no better than tossing a coin, to determine the presence or absence of deficiency. The study by Clarke et al.28 provides data which permits calculation of the specificity and sensitivity of a current B12 immunoassay in the detection of cobalamin deficiency in a community study of 1621 subjects over age 65 with normal renal function. Subjects were defined as cobalamin deficient if the methylmalonic acid was elevated above 0.75 μmol/l. Deficiency was found in 4.3% of subjects over 65 years of age with normal renal function. The mean B12 level of these subjects was 151 pmol/l (202 ng/l) by Siemens Centaur assay (range 110–199 pmol/l). Table 10.4 illustrates the calculation to derive specificity and sensitivity for the Siemens Centaur B12 assay using a cut-off point of 200 pmol/l (270 ng/l).
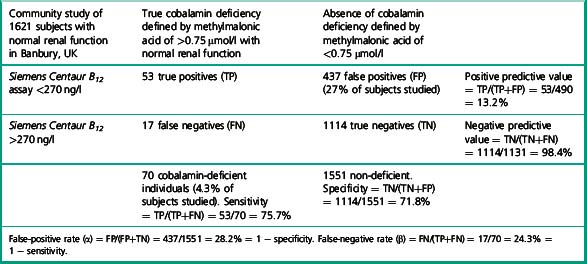
Selection of a cut-off point for cobalamin of below 150 ng/l, will identify subjects with higher probability of presence of cobalamin deficiency. Some authors13 have advocated choosing a cut-off point 25% below the reference range lower limit for any particular assay.
If, for example, a cut-off point of 125 pmol/l (168 ng/l) is chosen, the specificity of a value below this level, i.e. the number of normal individuals who fall below the cut-off point, will be markedly reduced (FP) and therefore the specificity of the test (TN/TN+FP) improves to 95%, although the detection of individuals with true deficiency who lie above the cut-off point, i.e. false negatives will be markedly increased, resulting in a sensitivity (TP/TP+FN) of 35%. The ROC curve therefore allows a laboratory to select a cut-off point that meets the objective of the laboratory – to have either a highly specific but low sensitivity test or to have a test of poor specificity but high sensitivity. Clearly cobalamin assays do not meet the criteria for an ideal test of high sensitivity and high specificity which would lie on the coordinates 0.1 to the left of the graph. The Axis-Shield/Abbott holotranscobalamin assay in this pivotal study is seen to have slightly superior ROC curves (see Fig. 10.4).
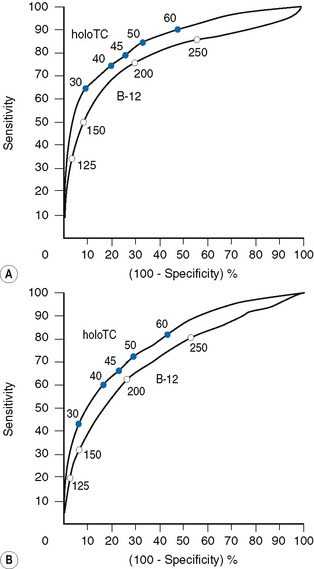
Utility of holotranscobalamin, methylmalonic acid and homocysteine assays
Holotranscobalamin assays gave a greater area under the curve, 0.85 versus 0.76 in the above study, and superior sensitivity and specificity. Holotranscobalamin is the physiologically active fraction of total cobalamin.27–29 An isolated abnormal result of serum B12 should not be the sole criterion on which treatment decisions are based and a repeat assay and other confirmatory and clinical evaluation are necessary prior to a diagnostic conclusion. Additional secondary testing with metabolite levels, or holotranscobalamin and monitoring of treatment response is recommended. High B12 levels have been described in subjects with no myeloproliferative neoplasm and who are not on cobalamin therapy or vitamin supplementation.35 This is thought to be due to immunoglobulin-complexed B12 resulting in assay interference. False normal B12 levels24,25 have been described in subjects with high titre intrinsic factor antibody25 and may also occur due to presence of heterophile antibody interference.
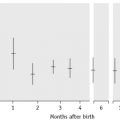
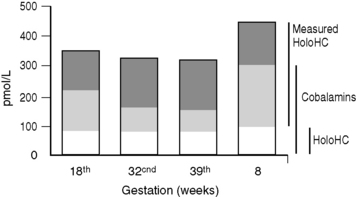
Holotranscobalamin assays27,28 may challenge total B12 assays as a first-line test in cobalamin assessment. Holotranscobalamin has been shown to be unaffected by assay interference from high-titre intrinsic factor antibody levels.36 In addition, holotranscobalamin is not subject to the 30% fall in total B12 levels seen in normal pregnancy, which makes low total B12 levels uninterpretable during pregnancy (Fig. 10.5).30
Access to homocysteine, methylmalonic acid and holotranscobalamin assays facilitates more precise definition of cobalamin and folate status in patients in whom prolonged B12 therapy may have been initiated inappropriately and continued unnecessarily or, conversely, discontinued inappropriately because of lack of confidence in the original assessment. The study by Gorringe et al.16 showed that only 27/49 patients were anaemic or macrocytic with a low total B12 of <170 ng/l and elevated MMA. All of these patients treated with B12 therapy corrected elevated MMA levels, suggesting the presence of a metabolic deficiency. However, only 15/27 showed a haematological response.
Homocysteine levels also fell by >25% in 47/49 patients after B12 treatment. Many of these patients had no clinical evidence or symptoms of cobalamin deficiency and may reflect subjects with subclinical deficiency, which could have subtle cognitive impairment, or may just represent a compensated metabolic state of no clinical consequence. Elevation of homocysteine levels is seen in folate deficiency and is therefore less specific than MMA measurement. The causes of cobalamin deficiency are shown in Table 10.5.
Table 10.5 The causes of cobalamin deficiency
| Supportive information/diagnostic tests | ||
|---|---|---|
| Reduced intake | ||
| Strict vegetarian/vegan | Dietary history | |
| Dietary fad that excludes dairy products and meat | Ethnic origin/culture | |
| Breastfed babies of mothers who are vegetarian or cobalamin deficient | ||
| Poor dietary intake in elderly | ||
| Malabsorption as a result of loss or inactivity of intrinsic factor | ||
| Addisonian pernicious anaemia | Diagnostic criteria for pernicious anaemia (see Table 10.3) | |
| Gastrectomy (partial or total) | History of gastric surgery | |
| Bacterial overgrowth or parasitic infestation of small bowel | Radiolabelled lactose breath tests for bacterial overgrowth Repeat Schilling test post-antibiotic therapy | |
| Pancreatic dysfunction: failure of trypsin release of B12 from R binding proteins | Pancreatic function tests; exocrine pancreatic dysfunction results in abnormal Schilling test but clinical deficiency is rare | |
| Malabsorption as a result of failure of B12-intrinsic factor complex uptake in ileum – ileal resection | ||