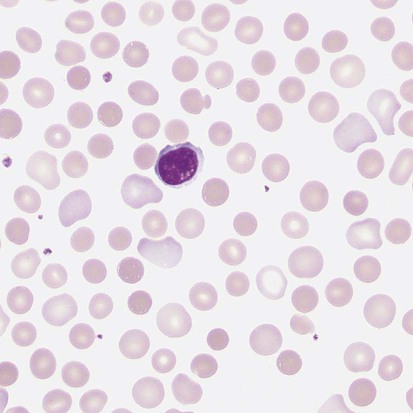
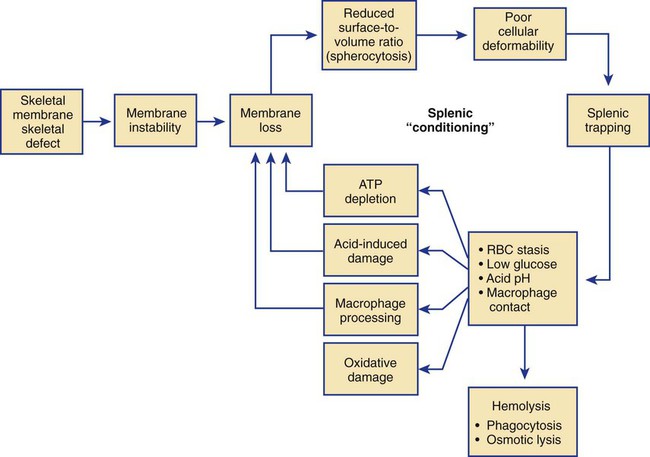
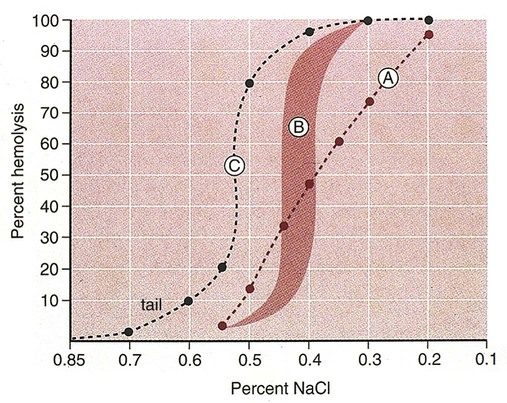
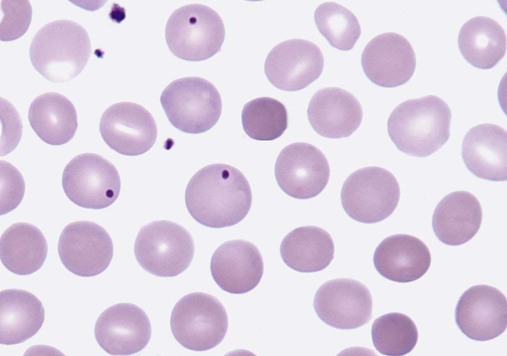
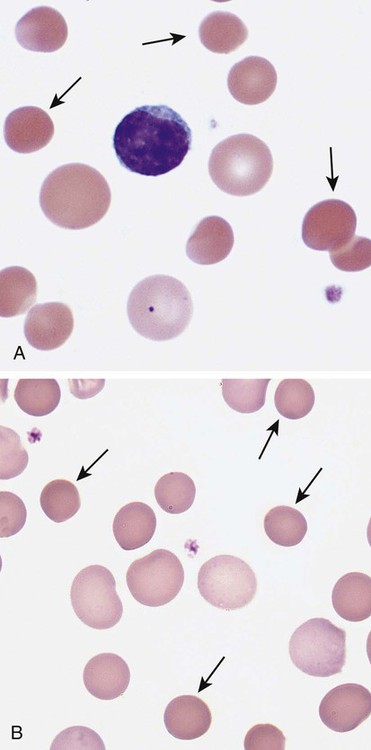
After completion of this chapter, the reader will be able to: 1. Describe the intrinsic cell properties that affect red blood cell (RBC) deformability. 2. Explain how defects in vertical and horizontal membrane protein interactions can result in a hemolytic anemia. 3. Compare and contrast the inheritance pattern, membrane proteins mutated, mechanism of hemolysis, typical RBC morphology, and clinical and laboratory findings of hereditary spherocytosis, hereditary elliptocytosis, and hereditary ovalocytosis. 4. Compare and contrast the RBC morphology and laboratory findings of hereditary spherocytosis and immune-associated hemolytic anemias. 5. Explain the principle, interpretation, and limitations of the osmotic fragility test in the diagnosis of hereditary spherocytosis. 6. Describe the causes, pathophysiology, RBC morphology, and clinical and laboratory findings of hemolytic anemias characterized by stomatocytosis. 7. Describe the causes and pathophysiology of hereditary and acquired conditions characterized by acanthocytosis. 8. Describe the cause, pathophysiology, clinical manifestations, laboratory findings, and treatment for paroxysmal nocturnal hemoglobinuria. 9. Compare and contrast the inheritance pattern, pathophysiology, clinical symptoms, and typical laboratory findings of glucose-6-phosphate dehydrogenase deficiency and pyruvate kinase deficiency. 10. Given the history, symptoms, laboratory findings, and a representative microscopic field from a peripheral blood film for a patient with a suspected intrinsic hemolytic anemia, discuss possible causes of the anemia and indicate the data that support these conclusions. The peripheral blood film revealed anisocytosis, polychromasia, and spherocytes (Figure 23-1). 1. From the data given, what is a likely cause of the anemia? 2. What additional laboratory tests would be of value in establishing the diagnosis, and what abnormalities in the results of these tests would be expected to confirm your impression? Intrinsic hemolytic anemias comprise a large group of disorders in which defects in the red blood cells (RBCs) themselves result in premature hemolysis and anemia. Intrinsic disorders can be divided into abnormalities of the RBC membrane, metabolic enzymes, or hemoglobin (Hb). Most of these defects are hereditary. This chapter covers defects in the RBC membrane and enzymes causing hemolytic anemia. Chapter 26 covers qualitative hemoglobin disorders, and Chapter 27 covers quantitative hemoglobin disorders. The RBC maintains a biconcave discoid shape that is essential for normal function and survival for 120 days in the peripheral circulation. The key to maintaining this shape is the plasma membrane, a lipid bilayer embedded with proteins and connected to an underlying protein skeleton (see Figure 9-2). The insoluble lipid portion serves as a barrier to separate the vastly different ion and metabolite concentrations of the interior of the RBC from its external environment, the blood plasma. The concentration of the constituents in the cytoplasm is tightly regulated by proteins embedded in the membrane that serve as pumps and channels for movement of ions and other material between the RBC’s interior and the blood plasma. Various membrane proteins also act as receptors, RBC antigens, enzymes, and support for the surface carbohydrates to form a protective glycocalyx with the surface glycolipids. The lipid bilayer remains intact because transmembrane proteins embedded in the membrane anchor it to a two-dimensional protein lattice (skeleton) immediately beneath its surface.1 Together, the transmembrane proteins and underlying skeleton provide structural integrity, cohesion, and mechanical stability to the cell. In a normal life span of 120 days, RBCs must repeatedly maneuver through very narrow capillaries and squeeze through the splenic sieve (narrow slits or fenestrations in the endothelial cell lining of the splenic sinuses) as they move from the splenic cords to the sinuses. To accomplish this without premature lysis, the RBCs must have deformability, or the ability to repeatedly bend, stretch, distort, and then return to the normal discoid, biconcave shape.2 The cellular properties that enable RBC deformability include (1) the RBC’s biconcave, discoid geometry, (2) the viscoelastic nature (elasticity) of its membrane, and (3) its cytoplasmic viscosity (see Chapter 9).3,4 The biconcave, discoid geometry of the RBC is dependent on vertical and horizontal interactions between the transmembrane and skeletal proteins (see Tables 9-5 and 9-6).3 Two transmembrane protein complexes, the ankyrin complex and protein 4.1 junctional complex, provide vertical structural integrity to the cell by anchoring the lipid bilayer to the underlying spectrin skeleton.3,5 In the ankyrin complex, ankyrin and protein 4.2 link transmembrane proteins, band 3, and RhAG (the Rh-associated glycoprotein) to the skeleton.6,7 In the protein 4.1 junctional complex, protein 4.1R links transmembrane proteins, glycophorin C, and XK, Rh, and Duffy proteins to the skeleton, and adducin and dematin link with transmembrane proteins, band 3, and glucose transport (glut 1) (see Figure 9-2).8 These interactions are called vertical because they are perpendicular to the plane of the cytoskeleton. They prevent loss of membrane and the resultant decrease in the surface area–to–volume ratio of the RBC.5 Two major skeletal proteins, α-spectrin and β-spectrin, interact laterally with each other to form antiparallel heterodimers, which link with other spectrin heterodimers to form tetramers. The spectrin heterodimers also form a spectrin–actin–protein 4.1R junctional complex with accessory proteins (tropomyosin, tropomodulin, dematin, and adducin), thus linking the spectrin tetramers in a two-dimensional lattice.9,10 These proteins provide horizontal mechanical stability, which prevents the membrane from fragmenting in response to mechanical stress.3,5 Factors that impact the elasticity of the cell are not as clear. Interactions between the spectrin dimers and the junctional complexes are flexible and allow for movement as the RBCs stretch and bend (see Figure 9-4).10 In addition, the ability of spectrin repeats to unfold and refold is likely to be one of the determinants of membrane elasticity.10,11 The cytoplasmic viscosity depends on the concentration of hemoglobin, as well as the maintenance of the proper cell volume by the normal functioning of various channels and pumps that allow the passage of ions, water, and macromolecules in and out of the cell.3 A defect in the RBCs that changes the membrane geometry, its elasticity, or the viscosity of the cytoplasm affects RBC deformability and can result in premature hemolysis and anemia.3 The RBC membrane is discussed in more detail in Chapter 9; the reader is encouraged to review that chapter when studying defects in the membrane. Most defects in the RBC membrane that can cause hemolytic anemia are hereditary; however, acquired defects also exist. Hereditary RBC membrane defects have historically been classified by morphologic features. The major disorders are hereditary spherocytosis, characterized by spherocytes, and hereditary elliptocytosis, characterized by elliptical RBCs. Hereditary pyropoikilocytosis, a variant of hereditary elliptocytosis, is characterized by marked poikilocytosis and heat sensitivity. Other less common membrane disorders include hereditary ovalocytosis, overhydrated hereditary stomatocytosis, and dehydrated hereditary stomatocytosis. Hereditary membrane defects can also be classified as those that affect membrane structure (and alter geometry and elasticity) and those that affect membrane transport (and alter cytoplasmic viscosity) (Box 23-1).10,12 The membrane structural defects can be further divided into those that affect vertical membrane protein interactions and those that affect horizontal membrane protein interactions.5,10 The major hereditary membrane defects are described in Table 23-1. TABLE 23-1 Characteristics of Major Hemolytic Anemias Caused by Hereditary Membrane Defects Hereditary spherocytosis (HS) is a heterogenous group of hemolytic anemias caused by defects in proteins that disrupt the vertical interactions between transmembrane proteins and the underlying protein cytoskeleton. HS has worldwide distribution and affects 1 in 3000 individuals of northern European ancestry.10 In 75% of families, it is inherited as an autosomal dominant trait and is expressed in heterozygotes who have one affected parent. Homozygotes are rare; such patients present with severe hemolytic anemia, but have asymptomatic parents.13 In approximately 25% of cases, the inheritance is nondominant, with some autosomal recessive cases.10,13 HS results from gene mutations in which the defective proteins disrupt the vertical linkages between the lipid bilayer and the skeletal network.5 Various mutations in six known genes can result in the HS phenotype (see Table 23-1). Mutations can occur in genes for (1) anchoring proteins, including ANK1, which codes for ankyrin (40% to 65% of cases in the United States and Europe; 5% to 10% of cases in Japan), and EPB42, which codes for protein 4.2 (fewer than 5% of cases in the United States and Europe; 45% to 50% of cases in Japan); (2) transmembrane proteins, including SLC4A1, which codes for band 3 (20% to 35% of cases), and RHAG, which codes for RhAG (fewer than 1% of cases); or (3) skeletal proteins, including SPTA1, which codes for α-spectrin (fewer than 5% of cases) and SPTB, which codes for β-spectrin (15% to 30% of cases).10,14 Because of the vertical interactions of the transmembrane proteins and skeletal protein lattice, a primary mutation in one gene may have a secondary effect on another protein in the membrane. For example, primary mutations in ANK1 result in both ankyrin and spectrin deficiencies in the RBC membrane.14,15 In approximately 10% of patients, no mutation is identified.10 The defects in vertical membrane protein interactions cause RBCs to lose unsupported lipid membrane over time because of local disconnections of the lipid bilayer and underlying skeleton. Essentially, small portions of the membrane peel off with little loss of volume. The RBCs acquire a decreased surface area–to–volume ratio, and the cells become spherical. These cells do not have the deformability of normal biconcave discoid RBCs, and their survival in the spleen is decreased.4,13,14 As the spherocytes attempt to move through the narrow, elliptical fenestrations of the endothelial cells lining the splenic sinusoids, they acquire further membrane loss or become trapped and are rapidly removed by the macrophages of the red pulp of the spleen.4,13 In addition, as the RBCs are sequestered in the spleen, the membrane can acquire yet more damage, lose more lipid membrane, and become more spherical due to splenic conditioning.4,13,14 The conditioning may be enhanced by the acidic conditions in the spleen and the prolonged contact of the RBCs with macrophages.13,14 Low levels of adenosine triphosphate (ATP) and glucose, and phagocyte-produced free radicals, which cause oxidative damage, may also play a role (Figure 23-2).14 In HS, RBC membranes also have abnormal permeability to cations, particularly sodium and potassium, which is likely due to disruption of the integrity of the protein skeleton.16 This results in a higher intracellular concentration of sodium and lower concentration of potassium compared with normal cells. The abnormality is not related to defects in cation transport proteins, and it occurs regardless of the type of primary mutation causing the HS.16 Symptomatic HS has three key clinical manifestations: anemia, jaundice, and splenomegaly. Symptoms of HS may first appear in infancy, childhood, or adulthood, or even at an advanced age. There is wide variation in symptoms. Silent carriers are clinically asymptomatic with normal laboratory findings and usually are identified only if they are the parents of a child with recessively inherited HS.4,13 Approximately 20% of patients have mild HS and are asymptomatic, because an increase in erythropoiesis compensates for the RBC loss.10,13 They usually have normal hemoglobin levels, but may show subtle signs of HS, with an increased reticulocyte count of up to 6% and a few spherocytes on the peripheral blood film.4,10,15 Mild HS may first become evident during pregnancy, during illnesses that cause splenomegaly (such as infectious mononucleosis), or in aging when the rate of erythropoiesis starts to decline.13,15 Approximately 60% of patients have moderate HS with incompletely compensated hemolysis, hemoglobin levels greater than 8 g/dL, reticulocyte counts greater than 6%, and spherocytes on the peripheral blood film.10,14,15 Jaundice is seen at some time in about half of these patients, usually during viral infections. About 10% of patients have moderately severe HS with hemoglobin levels usually in the range of 6 to 8 g/dL and reticulocyte counts greater than 10%.10,14 About 3% to 5% of patients have severe HS with hemoglobin levels below 6 g/dL and reticulocyte counts greater than 10%.10,14 Patients with severe HS are usually homozygous for HS mutations and require regular transfusions.10 Splenomegaly is found in about half of young children and 75% to 95% of older children and adults with HS.10 The hallmark of HS is spherocytes on the peripheral blood film. When present in patients with childhood hemolytic anemia and a family history of similar abnormalities, the uniform spherocytes are highly suggestive of HS. Some of these are microspherocytes—small, round, dense RBCs that are filled with hemoglobin and lack a central pallor (Figure 23-3). Normal-appearing RBCs, along with polychromasia and varying degrees of anisocytosis and poikilocytosis, are present. In addition to spherocytes, occasionally other RBC morphologic variants may be observed in some types of mutations: acanthocytes in some β-spectrin mutations,17 pincered or mushroom-shaped cells in some cases of band 3 deficiency in patients without splenectomy,18 ovalostomatocytes in homozygous EPB4.2 mutations,19 and stomatocytes in those with no expression of RHAG, the Rh-null phenotype.20 Note that spherocytes are not specific for HS and can be seen in other hereditary and acquired conditions. Because of the spherocytosis, HS patients have an increase in the mean cell hemoglobin concentration (MCHC) to between 35 and 38 mg/dL and an increase in the red cell distribution width (RDW) to greater than 14%.4,15 Automated hematology analyzers can detect the hyperdense RBCs found in HS.13 Biochemical evidence of extravascular hemolysis may be present in moderate to severe forms of HS, and the extent is dependent upon the severity of the hemolysis. This includes a decrease in serum haptoglobin level and an increase in serum indirect bilirubin level (see Chapter 22). The bone marrow shows erythroid hyperplasia due to the increased demand for RBCs to replace the circulating spherocytes that are prematurely destroyed, but bone marrow analysis is not required for diagnosis. In cases in which HS is suspected, but the family history and mode of inheritance are not clear, or there are atypical clinical and/or laboratory findings, further special testing is needed for diagnosis.15 The osmotic fragility test demonstrates increased RBC fragility in blood samples in which cells have decreased surface area–to–volume ratios. In this test, when RBCs are put into a series of increasingly hypotonic sodium chloride (NaCl) solutions, water enters the cells until equilibrium is achieved. As this phenomenon occurs, the cells swell until the internal volume is too great, which causes them to lyse. Because spherocytes already have a decreased surface area–to–volume ratio, they lyse in less hypotonic solutions than normal-shaped, biconcave RBCs and thus have increased osmotic fragility. A plot of an osmotic fragility curve is shown in Figure 23-4. Normal biconcave RBCs show initial hemolysis at 0.45% NaCl, and hemolysis is complete at 0.30% NaCl. If the curve is shifted to the left, the patient’s RBCs have increased osmotic fragility. Conversely, if the curve is shifted to the right, the RBCs have decreased osmotic fragility. Decreased osmotic fragility is found in conditions characterized by numerous target cells, such as thalassemia. The osmotic fragility test is a useful confirmatory test for HS; however, a major drawback is its lack of sensitivity and specificity.15 In terms of sensitivity, the test result can be normal in patients with HS, especially the mild forms, so a normal osmotic fragility test finding does not rule out HS.15 In patients with mild HS and a normal osmotic fragility test result, incubating the blood at 37° C for 24 hours before performing the test may enable its detection of mild disease. During this incubation period, HS cells tend to become more spherical. Patients who have increased osmotic fragility only when their blood is incubated tend to have a low number of spherocytes in the total RBC population. In the osmotic fragility test without incubation, the existence of a distinct subpopulation of the most fragile cells, those most conditioned by the spleen, is reflected by the presence of a “tail” on the osmotic fragility curve (see Figure 23-4).13 After splenectomy is performed, the osmotic fragility improves, and this subpopulation of conditioned cells disappears.13 The osmotic fragility test is also nonspecific. A result indicating increased fragility does not differentiate between HS and spherocytosis caused by other conditions, such as burns, immune hemolytic anemias, and other acquired disorders.13,15 The autohemolysis test is relatively sensitive to HS but is not routinely used in many laboratories because the test is time consuming. It probably has sensitivity similar to that of the osmotic fragility test with incubation.4,13 When normal RBCs are incubated in their own plasma, hemolysis gradually takes place. In HS, because of the loss of membrane and the inability to maintain the cation gradient, the hemolysis is much greater. Normal samples generally have less than 5.0% hemolysis at the end of the 48-hour incubation period, and they have less than 1.0% if a source of glucose is added. The glucose provides energy to drive the cation pumps to help maintain the cells. In HS, the hemolysis is 10% to 50%, which corrects considerably but not to the normal range when glucose is added. In an HS patient with numerous microspherocytes conditioned in vivo by the spleen, the hemolysis may not correct with glucose. In atypical HS cases additional tests may be required to identify the primary gene defect or to determine the inheritance pattern.15 Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) can be used to identify membrane protein deficiencies by electrophoretic separation of the various proteins in solubilized RBC membranes with quantitation of the proteins by densitometry.13,15 Membrane proteins can also be quantitated by radioimmunoassay.13 Variation in membrane surface area and cell water content can be determined by osmotic gradient ektacytometry.21 The ektacytometer is a laser-diffraction viscometer that records the laser diffraction pattern of a suspension of RBCs exposed to constant shear stress in solutions of varying osmolality from hypotonic to hypertonic. An RBC osmotic deformability index is calculated and plotted against the osmolality of the suspending solution to generate an osmotic gradient deformability profile.21 This method, however, is available only in specialized laboratories.12,13 Patient blood can be screened for HS after incubation with eosin-5′-maleimide (EMA) and measurement of the fluorescence in a flow cytometer. EMA binds to band 3 and Rh-related proteins in the RBC membrane, and RBCs from HS patients with deficiencies in these proteins have 25% to 30% lower fluorescence than RBCs from normal controls and from patients with spherocytes due to immune-mediated hemolysis.22 The hypertonic cryohemolysis test is based on the fact that cells from HS patients are particularly sensitive to cooling at 0° C in hypertonic solutions.23 The percentage of hemolysis is calculated after the patient’s RBCs are incubated in buffered 0.7 mol/L sucrose, first at 37° C for 10 minutes, then at 0° C for 10 minutes. Normal cells show 3% to 15% hemolysis, whereas RBCs in HS have greater than 20% hemolysis.23 This test is sensitive in detecting HS, but increased hemolysis can also occur in Southeast Asian ovalocytosis, some types of hereditary elliptocytosis, and congenital dyserythropoietic anemia type II.15,23 Molecular techniques for detection of genetic mutations can be difficult and costly to use because of the large size of the genes involved and the number of possible mutations.12,13 The polymerase chain reaction procedure followed by single-strand conformational polymorphism analysis can identify potential regions in HS genes that may contain a mutation.13,15 Once a region has been identified, nucleic acid sequencing can be performed to identify the specific mutation.13 Although most patients with HS have a well-compensated hemolysis and are rarely symptomatic, complications may occur that require medical intervention. Patients may experience various crises, classified as hemolytic, aplastic, and megaloblastic.13 Hemolytic crises are rare and usually associated with viral syndromes. In aplastic crises there is a dramatic decrease in hemoglobin level and reticulocyte count. The crisis usually occurs in conjunction with parvovirus B19 infection, which suppresses erythropoiesis, and patients can become rapidly and severely anemic, often requiring transfusion.15 This complication is more common in children, but can occur in adults.13,15 Patients with moderate and severe HS can also develop folic acid deficiency resulting from increased folate utilization to support the chronic erythroid hyperplasia in the bone marrow.13 This phenomenon is termed megaloblastic crisis and is particularly acute during pregnancy and during recovery from an aplastic crisis. Providing folic acid supplementation to patients with moderate and severe HS avoids this complication.15 About half of patients, even those with mild disease, also experience cholelithiasis (bilirubin stones in the gallbladder or bile ducts) due to the chronic hemolysis.13 Chronic ulceration or dermatitis of the legs is a rare complication.13 Mild HS usually requires no treatment.15 In moderate and severe cases, splenectomy prevents clinically significant hemolysis, results in longer RBC survival in the peripheral blood, and helps prevent gallstones by decreasing the amount of hemolysis and thus the amount of bilirubin produced.15 Patients with uncomplicated disease have an excellent response. The major drawback of splenectomy is the lifelong risk of overwhelming sepsis and death, particularly after infection with Streptococcus pneumoniae.13,15 Patients receive pneumococcal vaccination and penicillin prophylaxis to reduce the risk of sepsis, but this does not completely eliminate the risk.15 Because infants and young children are especially susceptible to postsplenectomy sepsis, splenectomy is usually postponed until after age 6 years.15 In a recent nationwide sample of 1657 children (aged 5 to 12 years) with HS who underwent splenectomy, there were no cases of postoperative sepsis and no fatalities from any cause during hospitalization for the surgery.24 More recently, partial splenectomy has been performed in young children with severe HS, but further evaluation of the risks and benefits of this procedure are needed.13 After splenectomy, spherocytes are still apparent on the blood film and the osmotic fragility is still increased, but conditioned microspherocytes are no longer present, as evidenced by the disappearance of the tail of the osmotic fragility curve. All of the typical changes in RBC morphology seen after splenectomy also are observed, including Howell-Jolly bodies, target cells, and Pappenheimer bodies (Figure 23-5). Reticulocyte counts decrease to high-normal levels, and the anemia is usually corrected. Leukocytosis and thrombocytosis are present. Bilirubin levels decrease but may remain in the high-reference range. Occasionally, a patient does not improve because of an accessory spleen missed during surgery or the accidental autotransplantation of splenic tissue during splenectomy, and hemolysis may resume years later.13 The resumption of splenic function can be ascertained from liver or spleen scans.13 Patients with severe HS usually require regular transfusions. Transfusions are rarely needed in the less severe forms of HS.14 HS must be distinguished from immune-related hemolytic anemia with spherocytes. Family history and evaluation of family members, including parents, siblings, and children of the patient, help differentiate the hereditary disease from the acquired disorder. The immune disorders with spherocytes are usually characterized by a positive result on the direct antiglobulin test (DAT), whereas the results are negative in HS. The osmotic fragility test is not diagnostic of HS, because the cells in acquired hemolytic anemia with spherocytes also show increased osmotic fragility. In cases with a family history of HS, typical physical findings, an increased reticulocyte count, spherocytes on the peripheral blood film, and a negative result on the DAT, no further special testing is needed for the diagnosis of HS.15 The typical clinical and laboratory findings in HS are summarized in Table 23-2. TABLE 23-2 Typical Clinical and Laboratory Findings in Hereditary Spherocytosis (HS) *Varies with severity of HS and ability of the bone marrow to compensate for the hemolysis. †With some rare mutations, acanthocytes, pincered cells, stomatocytes, or ovalocytes may be seen in addition to spherocytes. ‡Incubation of blood at 37° C for 24 hours prior to testing may be required to detect mild disease. A normal osmotic fragility test finding does not rule out HS. Adapted from Botlon-Maggs PHB, Stevens RF, Dodd NJ, et al: Guidelines for the diagnosis and management of hereditary spherocytosis, Br J Haematol 126:459, 2004. Hereditary elliptocytosis (HE) is a heterogenous group of hemolytic anemias caused by defects in proteins that disrupt the horizontal or lateral interactions in the protein cytoskeleton. It reportedly exists in all of its forms in 1 in 2000 to 1 in 4000 individuals, but because the majority of cases are asymptomatic, the actual incidence is not known.25 The disease is more common in Africa and Mediterranean regions where there is a high prevalence of malaria. The prevalence in West Africa of certain spectrin mutations associated with HE is between 0.6% and 1.6%.26 The molecular basis for the association of spherocytosis and malaria is unknown. The inheritance pattern in HE is autosomal dominant.10 HE results from gene mutations in which the defective proteins disrupt the horizontal linkages in the protein cytoskeleton and weaken the mechanical stability of the membrane.5 The HE phenotype can result from various mutations in at least three genes: SPTA1, which codes for α-spectrin (65% of cases); SPTB, which codes for β-spectrin (30% of cases); and EPB41, which codes for protein 4.1R (5% of cases) (see Table 23-1).10,25 The spectrin mutations disrupt spectrin dimer interactions and the EPB41 mutations result in weakened spectrin–actin–protein 4.1R junctional complexes.10 RBCs are biconcave and discoid at first, but become elliptical over time after repeated exposure to the shear stresses in the peripheral circulation. The extent of the disruption of the spectrin dimer interactions seems to be associated with the severity of the clinical manifestations.10 In severe cases, the protein skeleton is weakened to such a point that cell fragmentation occurs. As a result, there is membrane loss and a decrease in surface area–to–volume ratio that reduces the deformability of the RBCs. The damaged RBCs become trapped or acquire further damage in the spleen, which results in extravascular hemolysis and anemia. In general, patients who are heterozygous for a mutation are asymptomatic and their RBCs have a normal life span; those who are homozygous for a mutation or compound heterozygous for two mutations can have moderate to severe anemia that can be life-threatening.10 The RBCs in HE patients all show some degree of decreased thermal stability. Cases in which the RBCs show marked RBC fragmentation upon heating were previously classified as hereditary pyropoikilocytosis (HPP). HPP is now considered a severe form of HE that exists in either the homozygous or compound heterozygous state.10 Patients with the Leach phenotype lack the Gerbich antigens, glycoprotein C (GPC), and glycoprotein D (GPD).4
Intrinsic Defects Leading to Increased Erythrocyte Destruction
Case Study
Patient Results
Reference Range
WBCs (×109/L)
13.4
4.5-11.5
RBCs (×1012/L)
4.20
4.60-6.00
Hb (g/dL)
11.9
14.0-18.0
Hct (%)
32.4
40-54
MCV (fL)
77
80-100
MCH (pg)
28.3
26-32
MCHC (g/dL)
36.7
32-36
RDW (%)
22.9
11.5-14.5
Platelets (×109/L)
290
150-450
Red Blood Cell Membrane Abnormalities
Red Blood Cell Membrane Structure and Function
Hereditary Red Blood Cell Membrane Abnormalities
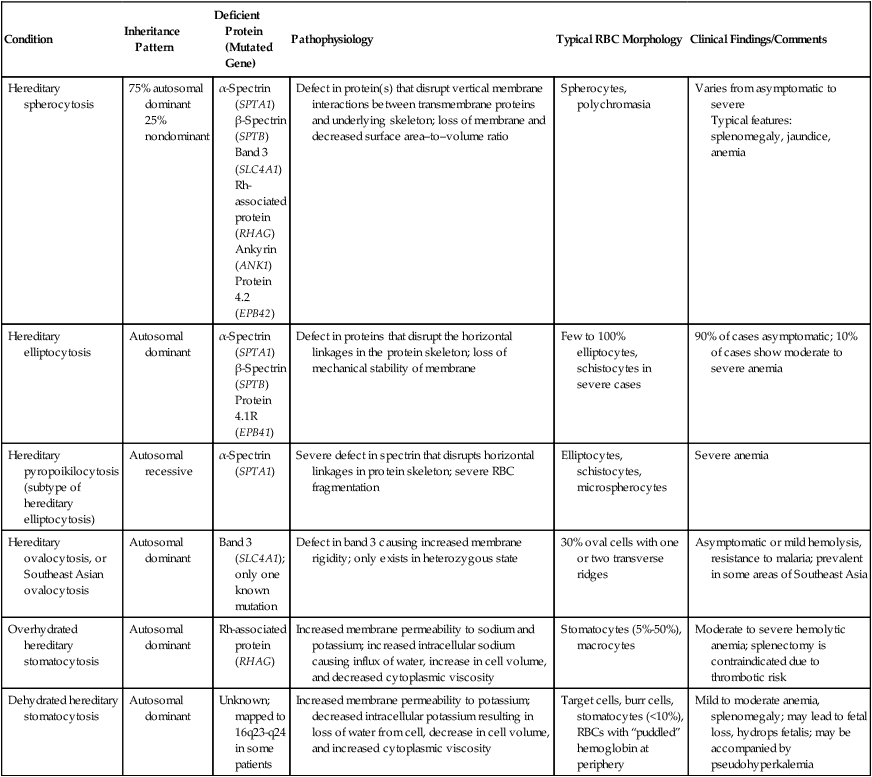
Condition
Inheritance Pattern
Deficient Protein (Mutated Gene)
Pathophysiology
Typical RBC Morphology
Clinical Findings/Comments
Hereditary spherocytosis
75% autosomal dominant
25% nondominant
α-Spectrin (SPTA1)
β-Spectrin (SPTB)
Band 3 (SLC4A1)
Rh-associated protein (RHAG)
Ankyrin (ANK1)
Protein 4.2 (EPB42)
Defect in protein(s) that disrupt vertical membrane interactions between transmembrane proteins and underlying skeleton; loss of membrane and decreased surface area–to–volume ratio
Spherocytes, polychromasia
Varies from asymptomatic to severe
Typical features: splenomegaly, jaundice, anemia
Hereditary elliptocytosis
Autosomal dominant
α-Spectrin (SPTA1)
β-Spectrin (SPTB)
Protein 4.1R (EPB41)
Defect in proteins that disrupt the horizontal linkages in the protein skeleton; loss of mechanical stability of membrane
Few to 100% elliptocytes, schistocytes in severe cases
90% of cases asymptomatic; 10% of cases show moderate to severe anemia
Hereditary pyropoikilocytosis (subtype of hereditary elliptocytosis)
Autosomal recessive
α-Spectrin (SPTA1)
Severe defect in spectrin that disrupts horizontal linkages in protein skeleton; severe RBC fragmentation
Elliptocytes, schistocytes, microspherocytes
Severe anemia
Hereditary ovalocytosis, or Southeast Asian ovalocytosis
Autosomal dominant
Band 3 (SLC4A1); only one known mutation
Defect in band 3 causing increased membrane rigidity; only exists in heterozygous state
30% oval cells with one or two transverse ridges
Asymptomatic or mild hemolysis, resistance to malaria; prevalent in some areas of Southeast Asia
Overhydrated hereditary stomatocytosis
Autosomal dominant
Rh-associated protein (RHAG)
Increased membrane permeability to sodium and potassium; increased intracellular sodium causing influx of water, increase in cell volume, and decreased cytoplasmic viscosity
Stomatocytes (5%-50%), macrocytes
Moderate to severe hemolytic anemia; splenectomy is contraindicated due to thrombotic risk
Dehydrated hereditary stomatocytosis
Autosomal dominant
Unknown; mapped to 16q23-q24 in some patients
Increased membrane permeability to potassium; decreased intracellular potassium resulting in loss of water from cell, decrease in cell volume, and increased cytoplasmic viscosity
Target cells, burr cells, stomatocytes (<10%), RBCs with “puddled” hemoglobin at periphery
Mild to moderate anemia, splenomegaly; may lead to fetal loss, hydrops fetalis; may be accompanied by pseudohyperkalemia
Mutations That Alter Membrane Structure
Hereditary Spherocytosis
Pathophysiology
Clinical and laboratory findings
Special tests
Complications
Treatment
Differential diagnosis
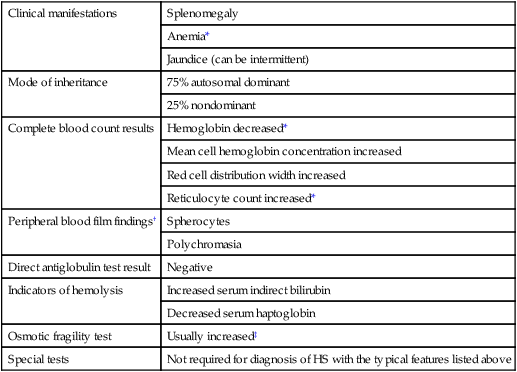
Clinical manifestations
Splenomegaly
Anemia*
Jaundice (can be intermittent)
Mode of inheritance
75% autosomal dominant
25% nondominant
Complete blood count results
Hemoglobin decreased*
Mean cell hemoglobin concentration increased
Red cell distribution width increased
Reticulocyte count increased*
Peripheral blood film findings†
Spherocytes
Polychromasia
Direct antiglobulin test result
Negative
Indicators of hemolysis
Increased serum indirect bilirubin
Decreased serum haptoglobin
Osmotic fragility test
Usually increased‡
Special tests
Not required for diagnosis of HS with the typical features listed above
Hereditary Elliptocytosis
Pathophysiology
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree