Chapter 27 Integrase Inhibitors and Other New Drugs in Development
INTRODUCTION
Currently approved ARV agents target one of four steps in the HIV-1 replicative cycle: viral entry, reverse transcriptase, protease or integrase activity. New agents are being developed that target these sites as well as other sites in the HIV-1 replicative cycle. These agents can be classified as inhibitors of viral entry, intracellular replication or assembly of infectious virions (Fig. 27-1). Inhibitors of viral entry include the CCR5 chemokine co-receptor antagonist, vicriviroc, which is in advanced stages of development as well as other attachment inhibitors and novel fusion inhibitors. While next-generation nucleoside and non-nucleoside reverse transcriptase inhibitors (NNRTIs) are being tested, HIV integrase inhibitors are the most promising class of compounds being developed to inhibit intracellular replication. Agents that inhibit viral assembly that are under development include novel PIs and maturation inhibitors. Our discussion will begin with a review of integrase inhibitors and will then move to other compounds that inhibit intracellular replication, viral entry and viral assembly.
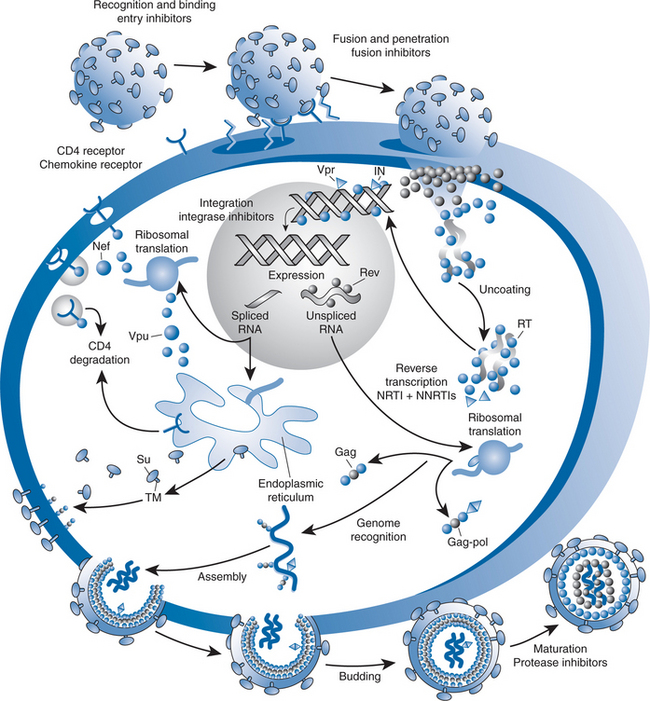
The information reported here summarizes available data on drugs that are in clinical trials, as well as some novel drugs that are in preclinical development (Table 27-1), and is not intended to be encyclopedic. Several new compounds in advanced stages of development are covered in more detail elsewhere in this text, including co-receptor inhibitors (see Chapter 24) and the PI darunavir (see Chapter 26). In addition, immune-based approaches and therapeutic vaccines are covered elsewhere (see Chapters 30 and 31 respectively).
Table 27-1 Selected Antiretroviral Agents in Development
| Agent | Manufacturer |
|---|---|
| Integrase Inhibitors | |
| Raltegravir (MK-0518) | Merck |
| Elvitegravir (GS 9137, JTK-303) | Gilead |
| Nucleoside Reverse Transcriptase Inhibitors | |
| D-d4FC (Reverset, DPC 817, dexelvucitabine) | Incyte |
| Elvucitabine (ACH-126,433, β-L-d4FC) | Achillion |
| Apricitabine (AVX-754, SPD-754, BCH-10618, (-)dOTC) | Avexa |
| Racivir (RCV; (+/−)-FTC; [(+/−)-β-2’,3’-dideoxy-5fluoro-3′-thiacytidine] | Pharmasset |
| Nonnucleoside Reverse Transcriptase Inhibitors | |
| Etravirine (TMC 125, R165335) | Tibotec/J&J |
| Rilpivirine (TMC 278, R278474) | Tibotec/J&J |
| GW678248/GW695634 | GlaxoSmithKline |
| Attachment Inhibitors | |
| PRO-542 | Progenics |
| Pharmaceuticals | |
| TNX-355 | Tanox/Biogen |
| BMS-488043 | Bristol-Myers |
| Squibb | |
| Fusion Inhibitors | |
| TR-290999 and TR291144 | Trimeris |
| Co-receptor Inhibitors | |
| See Chapter 24 | |
| Protease Inhibitors | |
| Brecanavir (GW640385) | GlaxoSmithKline |
| Maturation Inhibitors | |
| Bevirimat (PA-457) | Panacos |
INHIBITORS OF INTRACELLULAR REPLICATION
Integrase Inhibitors
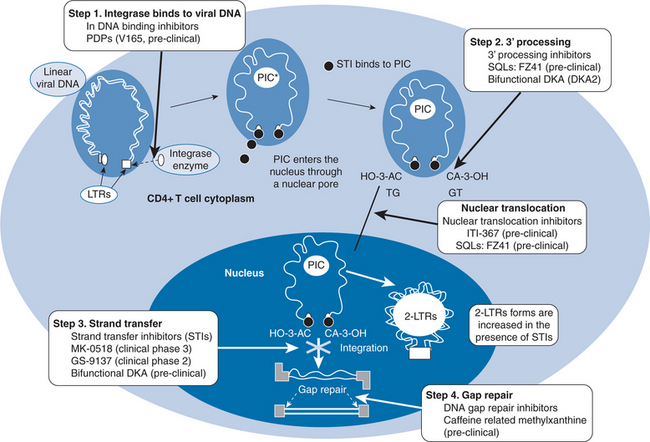
Integration of HIV-1 genetic material into host DNA is essential for productive infection with HIV-1.1 Inhibition of this process has been an attractive target for drug development since the identification of HIV-1 integrase in the 1980s. The mechanism by which integration occurs has been elucidated, and several inhibitors are in advanced stages of development. After HIV-1 enters target cells, viral reverse transcriptase (RT) synthesizes a linear, double-stranded DNA copy (cDNA). Insertion of viral cDNA into the chromosomal DNA of target cells is the hallmark of retroviral infection and is a prerequisite for transcription of the HIV-1 genome and translation of viral proteins.2,3 This process is catalyzed by the viral enzyme, integrase, which is a 288 amino acid protein derived from the 3′ end of the HIV polymerase gene. Integration of viral cDNA into host chromosomes is a multistep process that has both cytoplasmic and nuclear stages (Fig. 27-2).4 Following reverse transcription, HIV-1 cDNA binds to integrase in the cytoplasm via interactions with specific sequences in the HIV-1 LTR region. The binding of HIV-1 cDNA to integrase catalyzes the removal of two nucleotides proximal to each 3′-end of proviral cDNA in the cytoplasm of target cells (3′-processing). After 3′-processing, integrase remains bound to HIV-1 cDNA, forming multimeric pre-integration complexes (PICs). PICs interact with a variety of viral and cellular proteins to gain entry into the host nucleus. Once inside the nucleus, HIV-1 integrase catalyses insertion of viral cDNA into the host chromosome (strand transfer). Upon completion of the strand transfer process, the defects in the host cell’s genome that occur during cDNA insertion are repaired. Because there is no host equivalent, HIV-1 integrase has long been recognized as a potentially attractive target for ART.5 The development of in vitro assays that specifically evaluate the various steps in cDNA integration has been critical to integrase inhibitor development.6–8 Although many compounds have been reported to inhibit its activity, development of integrase inhibitors has been plagued by difficulties identifying compounds that specifically attack this target.9 At least four criteria need to be met in order to definitively establish that a compound targets integrase.4 First, it should be active during the replication cycle in which integration occurs (4–16 h after infection). Second, infected cells treated with the agent should show accumulation of circular cDNA. Third, mutations in integrase should be found in resistant viruses and finally, recombinant viruses into which mutant sequences of integrase are inserted should also be resistant to the putative inhibitor. A phenotypic assay of integrase activity has been developed to identify susceptibility of integrase inhibitors using recombinant viruses that express integrase alone or in combination with protease and reverse transcriptase.10 While lead compounds targeting each step in the process of integration into host DNA have been identified, the most promising agents currently being studied all inhibit the strand-transfer process and two different classes of strand-transfer integrase inhibitors will be reviewed here.
DKA Derivatives
Diketobutanoic acid derivatives (DKAs) were identified when molecules were screened for antiviral activity from a library of over 250 000 candidate compounds using an in vitro model of HIV-1 integration.8 Structural analysis of these substances indicated a common diketo acid moiety, thus leading to their classification as DKAs. DKAs inhibit HIV-1 cDNA integrase strand-transfer function at nanomolar to micromolar concentrations.8 The DKA L-870812 was the first integrase inhibitor to demonstrate in vivo antiviral activity in a simian human immunodeficiency virus (SHIV)-rhesus macaque model.11 Because of their structural similarities, agents within this class have induced both novel integrase mutations and the potential for cross-resistance.12,13 Biochemical modifications have improved the potency of the first generation of DKAs and second generation compounds of this class are at the leading edge of integrase inhibitor development. First generation compounds such as S-136014–18 and L-870,81019–24 demonstrated potent in vitro activity against HIV-1 but were not pursued because of either poor bioavailability (S–1360) or hepatotoxicity in animal models (L-870,810). These DKA derivatives, however, provided proof that integrase function could be inhibited and paved the way for development of other drugs in this promising new class of compounds.
Raltegravir (MK-0518)
Raltegravir is an evolutionary descendant of the diketobutanoic acids which its developers further classify as a pyrimidinone derivative. Raltegravir has demonstrated potent in vitro activity against wild-type HIV-1 (IC90 of 33 nM ± 23 nM in 50% human serum).25 Raltegravir maintains activity against HIV-1 regardless of virus co-receptor tropism, and is active against multidrug resistant isolates.26 The activity of raltegravir against nonclade B HIV-1 and against HIV-2 has not been reported. Isolates that developed in vitro resistance to raltegravir during serial passage studies retained susceptibility to all other existing classes of ARVs. A variety of naturally occurring polymorphisms within the HIV-1 integrase gene have been identified, but mutations that confer high-level resistance to integrase inhibitors in vitro include T66I/O, L74M, F121Y, T125K, G140S, S230R, V249I, and C280Y. These polymorphisms have not been shown to arise spontaneously without prior exposure to integrase inhibitors.27 Studies of virologic failure in subjects receiving raltegravir have demonstrated two common genetic pathways: N155H and Q148K/R/H. Additional mutations were observed in both pathways including E92Q, V151I, T97A, G163R, L74M within the N155H pathway and G140S/A, E138K within the Q148K/R/H pathway. Mutations are proximal to the catalytic center and are similar to those selected by passage of virus in the presence of drug in vitro. Raltegravir is metabolized by glucuronidation via UGT1A1 and does not induce or inhibit CYP3A4. Raltegravir has in vitro synergy with all currently available classes of ARV agents.25
Phase 1 studies suggested antiviral activity at a dose range between 100 and 800 mg twice daily, without regard to food intake. A phase II trial of raltegravir monotherapy assessed the antiviral activity, pharmacokinetics and tolerability in 35 treatment-naive, HIV-1-infected subjects.25 Subjects with an HIV-1 load >5000 copies/mL and a CD4 count of >100 cells/mm3 were randomized 4:1 to receive either 100, 200, 400, or 600 mg of raltegravir twice daily for 10 days versus placebo. HIV-1 load declined between 1.7 and 2.2 log10 copies/mL at the end of the study period and between 50% and 57% of treated subjects were able to achieve levels <400 copies/mL and 13–29% <50 copies/mL.28 Pharmacokinetic analysis at the end of the study demonstrated minimum concentration (Cmin) values for all doses studied that were greater than raltegravir’s IC90 in vitro. No serious adverse events (SAEs) or grade 3 or 4 laboratory abnormalities were observed.
Interim 16-week results from a study of the antiviral activity of raltegravir in treatment-experienced subjects have been reported.26 One hundred and sixty seven subjects with documented triple-class resistance, CD4 counts >100 cells/mm3 and plasma HIV-1 RNA levels of >5000 copies/mL were enrolled in a trial of optimized background therapy (OBT) with raltegravir versus placebo. Study subjects were highly treatment-experienced with ∼90% resistant to all avail-able PIs and ∼50% resistant to all available ARV agents, as measured by phenotyic sensitivity score (PSS). Subjects received 200, 400, or 600 mg of raltegravir versus placebo twice daily. At week 16, 65–85% of subjects receiving raltegravir had achieved an HIV-1 load <400 copies/mL and between 56% and 72% had <50 copies/mL. This compared to rates less than 20% in the OBT with placebo groups. Viral load decline was over 2 log10 copies/mL for all three of the arms receiving raltegravir, compared to almost 1 log10 copies/mL for those receiving OBT with placebo. CD4 counts increased more in the 400 mg and 600 mg raltegravir arms (100–110 cells/mm3), compared to either the 200 mg or placebo arms (∼30 cells/mm3). Adverse event rates were comparable between the groups receiving raltegravir and placebo, and were generally mild to moderate. Twenty-four week data have confirmed sustained responses and have highlighted the importance of using raltegravir with other active agents.29 Efficacy of raltegravir in achieving HIV-1 RNA level <400 copies/mL rose from 54–69% in participants with a PSS score of 0, to 75–84% in those with a PSS of 1-2. Similarly, addition of raltegravir to OBT with enfuvirtide in subjects who were fusion inhibitor-naive resulted in HIV-1 levels <400 copies/mL in over 90% of individuals at 24 weeks. Drug-related SAEs were reported in four subjects (pancreatitis, lacunar infarct, lipoatrophy, hepatomegaly, and metabolic acidosis, renal insufficiency and death). The first two SAEs were not attributable to raltegravir (pancreatitis was attributed to an agent in OBT and the lacunar infarct occurred in the placebo arm), but the relationship of the other SAEs to the study drug is uncertain as subjects and investigators remain blinded to the arm of the study in which they occurred. Diarrhea, nausea, fatigue, headache, and pruritus were seen at low rates in most arms but were comparable to those seen in the placebo arm. Because atazanavir is known to be an inhibitor of UGT1A1, a subgroup analysis is pending to study the potential interaction between these two compounds.
Preliminary data from two large multinational phase III trials of raltegravir have been presented.30,31 The studies known as BENCHMRK-1 and BENCHMRK-2 (blocking integrase in treatment-experienced patients with a novel compound against HIV: Merck) were designed to evaluate virologic and immunologic responses, as well as safety and tolerability of raltegravir versus placebo, in subjects with triple-class resistant HIV-1 infection who were receiving OBT. Subjects were required to have genotypic or phenotypic evidence of resistance to one drug in the NNRTI, PI and nucleoside reverse transcriptase inhibitor (NRTI) classes along with an HIV-1 RNA level of >1000 copies/mL. OBT was investigator selected and could include investigational compounds. Individuals qualifying for inclusion were randomized 2:1 to receive 400 mg bid of raltegravir versus placebo. Persons failing virologically (defined as <1 log10 copies/mL or >400 copies/mL or rebound viremia after achieving an undetectable plasma HIV RNA) at week 16 were allowed to enter an open-label raltegravir arm. BENCHMRK-1 enrolled 350 patients in Europe, Asia and the Pacific, while BENCHMRK-2 recruited 349 participants in North America, Central America and South America. Subjects in both trials were predominantly male (84–91%) and averaged 44–46 years of age. Subjects in BENCHMRK-1 were predominantly Caucasian (75–81%) compared to BENCHMRK-2 (55–65%). Over 90% of subjects had AIDS, and mean CD4 counts at entry ranged from 146 to 163 cells/mm3. Subjects were highly treatment-experienced with genotypic sensitivity score (GSS) of 0/1 in 30–33% of subjects receiving raltegravir in BENCHMRK-1 and 20–44% in BENCHMRK-2, compared to 29–41% and 26–40% for the placebo-arms of these two groups. About 20% of subjects were enfuvirtide-naive in both studies. About 27% of individuals in BENCHMRK-1 and 45% in BENCHMRK-2 were darunavir-naive compared to 25% and 50% in the placebo arms of both these trials, respectively. The 16-week data demonstrated that 77% of raltegravir-treated subjects achieved an HIV-1 level <400 copies/mL, compared to 41% and 43% in the placebo arms of BENCHMRK-1 and BENCHMRK-2, respectively. Patients receiving raltegravir demonstrated higher rates of suppression below 50 copies/mL (61% in BENCHMRK-1 and 62% in BENCHMRK-2, compared to 33% and 36% in the respective placebo arms. CD4 count rise averaged 83 cells/mm3 in the raltegravir arm of BENCHMRK-1 and 86 cells/mm3 in BENCHMRK-2, compared to rises of 31 and 40 cells/mm3 in the respective placebo arms. Not surprisingly, the efficacy of raltegravir was enhanced when the OBT contained one or more additional drug with anti-HIV activity. Although subjects receiving raltegravir achieved higher rates of virologic control than those receiving placebo, the effect was magnified in those with low GSS scores or in those who were naive to either enfuvirtide or darunavir. For example, 90% of subjects receiving raltegravir who were naive to darunavir but enfuvirtide-experienced achieved an HIV-1 RNA level of <400 copies/mL compared to 55% in those with identical darunavir and enfuvirtide experience who received placebo. Like most salvage trials, adverse events (AEs) were common in all arms of the trial (81% in the BENCHMRK-1/2 arms versus 83–86% in the placebo arms). Drug-related AEs (defined as possibly, probably or definitely-related to raltegravir, placebo or OBT drug) were also common (44% in BENCHMRK-1 and 53% in BENCHMRK-2 compared to51% and 52% in the placebo arms, respectively). Serious drug-related AEs were rare (<2.5% in all arms) as were AEs leading to discontinuation of study drugs (<3.4%). Adverse clinical experiences included gastrointestinal upset, injection site reaction, headache and fatigue, and were less frequent in the raltegravir arms of the study. Although not seen in the BENCHMRK-1 study, the raltegravir arms in BENCHMRK-2 appeared to have slightly higher rates of specific gastrointestinal disturbances such as diarrhea, abdominal distension/pain and flatulence, compared to those in the placebo arm. Grade 3–4 laboratory abnormalities were uncommon and were not more common than placebo in either trial.
The success of raltegravir in treatment-experienced patients has led the drug’s developers to initiate a phase III trial comparing raltegravir to efavirenz in treatment-naive patients.32 24-week results from this trial have been reported.32 The study enrolled subjects from the previous 10-day monotherapy study by adding fixed-dosed tenofovir/lamivudine to the existing bid 600, 400, 200, and 100 mg arms of raltegravir, while replacing the placebo arm with efavirenz 600 mg once daily with tenofovir and emtricitabine. Approximately 40 patients were randomized to each of the five arms in this study. Subjects were enrolled in this trial if they had no prior ART and had a CD4 count of >100 cells/mm3 and an HIV-1 load >5000 copies/mL, with documented susceptibility to efavirenz, lamivudine and tenofovir on genotypic analysis. The study was designed to rule out non-equivalence of raltegravir-based ART compared to an efavirenz-based regimen, and assessed HIV-1 load responses, changes in CD4 cell count and safety/tolerability. Participants were primarily male (73-90%) and nonwhite (65–82%) and had comparable HIV-1 RNA levels and CD4 cell counts at entry. Rates of AIDS diagnosis varied slightly from 29% in the 400 mg arm to 43% in the 600 mg raltegravir arm. Thirty-seven percent of subjects in the efavirenz arm had AIDS at enrollment. At planned 24-week interim analysis, all four raltegravir arms demonstrated excellent virologic responses with the three highest dose arms all having plasma HIV-1 RNA levels <50 copies/mL in over 90% of subjects. The 100 mg-arm produced HIV-1 RNA levels <50 copies/mL in 87% of subjects. All arms were comparable to the 90% rates of HIV-RNA <50 copies/mL seen in the efavirenz arm. Although the clinical significance is uncertain, all four raltegravir arms had more rapid reductions of HIV-1 RNA levels compared to efavirenz, although by 24 weeks both raltegravir and efavirenz produced similar rates of virologic control. CD4 increases were comparable across arms and ranged from ∼100 cells/mm3 in the efavirenz arm to ∼175 cells/mm3 in the 100 mg raltegravir arm. Discontinuation rates were low in all arms. Side-effects were generally mild and comparable across all arms with the exceptions of headache, dizziness, and abnormal dreams, which were all more common in the efavirenz-treated subjects. SAEs were infrequent (4% in combined raltegravir group vs 3% in efavirenz arm) and none was felt to be drug related. Grade 3 or 4 laboratory abnormalities were uncommon. An analysis of the impact of raltegravir on total cholesterol and triglyceride levels in this population has been reported.33 No change was observed in either total cholesterol or triglyceride levels in any of the raltegravir arms compared to 19 mg/dL and 47 mg/dL rises, respectively, in the efavirenz arm. This trial continues to accrue patients and additional data on efficacy, safety, toxicity and pharmacology will be forthcoming.
Next-Generation DKAs
Utilizing detailed information on structure–activity relationships in a series of viral isolates with in vitro resistance to various strand-transfer integrase inhibitors (including raltegravir), a series of next-generation integrase inhibitors have been identified. These compounds were derived by rational modification of a diketo acid pharmacophore to create compounds that bind tightly to the catalytic core of HIV-1 integrase, despite the fact that it contains these integrase mutations. The most promising candidate next-generation integrase inhibitor, MK-2048, retained activity against three of the four types of drug-resistant integrase mutants. The remaining viral mutant, containing integrase mutations at E138, G140, and Q148, was somewhat resistant to MK-2048, but much more resistant to current lead compounds such as raltegravir or elvitegravir. MK-2048 has demonstrated excellent in vitro antiviral activity with an IC95 of 41 nM and has demonstrated favorable pharmacokinetics in dogs and rats. Further studies of MK-2048are planned.34
Quinoline Derivatives
The recognition that divalent cations were essential to the catalytic activity of HIV-1 integrase led to the identification of quinoline-derived compounds as a potential class of integrase inhibitors.35,36 The exact mechanism by which quinoline derivatives inhibit HIV-1 integrase activity is uncertain. Studies of styrylquinoline derivatives (SQLs) suggest that they interfere with both the cytoplasmic binding of integrase to viral cDNA and the intranuclear binding to target DNA prior to strand transfer.37 SQLs may also inhibit nuclear import of PICs.38 SQLs inhibit replication of HIV-1 at micromolarconcentrations in vitro.35
Elvitegravir (GS 9137, JTK-303)
Elvitegravir is a dihydroquinoline carboxylic acid that was derived from a quinolone antibiotic.39 Although quinolones have no activity against HIV-1, they do share common structural motifs with DKAs, a fact that led developers to study the activity of these compounds against HIV-1 integrase. Elvitegravir inhibits the strand transfer activity of HIV-1 integrase with an IC50 of 8.8 nM.40 Elvitegravir demonstrated in vitro activity against a variety of HIV-1 subtypes, as well as against a panel of drug-resistant clinical isolates (EC50 from 0.02 to 1.26 nM); elvitegravir is also active against HIV-2.41 In vitro combination studies suggest that elvitegravir is synergistic or additive with available ARVs.40 Serial passage studies of HIV-1 in the presence of elvitegravir have revealed two common pathways for the emergence of resistance in vitro, T66I and E92Q.42 Important differences exist between these two pathways in that the E92Q polymorphism confers cross-resistance in vitro to raltegravir while the T66I results in reduced susceptibility to elvitegravir alone. A variety of secondary mutations in integrase emerge with serial passages that further reduce sensitivity to elvitegravir, including H51Y, F121Y, S147G, S153Y, E157Q, and R263K. Isolates with multiple integrase-associated mutations created through site-directed mutagenesis retained in vitro susceptibility to tenofovir, lopinavir/ritonavir and efavirenz.
The safety and pharmacokinetics of elvitegravir have been studied in uninfected male volunteers.43 Thirty two subjects were randomized 3:1 to receive a single 100, 200, 400, or 800 mg dose of elvitegravir or placebo. The 400 mg group received a second dose with breakfast after an appropriate wash-out period to assess the effect of food on bioavailabilty. Maximum concentrations (Cmax) and area under the concentration curve (AUC) increased as dose increased and plasma concentrations exceeded the EC90 at 12–24 h. Elvitegravir’s half-life (T½) was 3 h. Administration of elvitegravir with food increased absorption threefold. Elvitegravir is metabolized via the cytochrome P450 system; administration of elvitegravir with 100 mg of ritonavir increases AUC ∼30-fold and to increase T½ to ∼9 h.44 No SAEs or grade 3 or 4 laboratory abnormalities were seen in this trial.
A phase 2 study of the antiviral activity, safety and tolerability of elvitegravir in HIV-1-infected persons has been reported.44 This prospective, randomized, double-blind, placebo-controlled, monotherapy trial enrolled 40 HIV-1-infected individuals and randomized them to receive either 200, 400, or 800 mg of elvitegravir twice daily, 800 mg of elvitegravir once daily, or 50 mg of elvitegravir once daily with 100 mg of ritonavir, versus placebo for 10 days. Subjects were required to be off all ART and to have a CD4 count of ≥200 cells/mm3 and an HIV-1 load between 10 000 and 300 000 copies/mL. Declines in HIV-1 load ranged from 0.89 log10 copies/mL for the 800 mg daily group to 2.03 log10 copies/mL for the ritonavir-boosted 50 mg group, compared to a decline of 0.13 log10 copies/mL seen in the placebo arm of the study. Adverse events were comparable between elvitegravir and placebo recipients, and were generally mild. The only adverse event that occurred with greater frequency in the elvitegravir groups was fatigue. Only two grade 3 laboratory abnormalities were reported in subjects receiving elvitegravir (hypertriglyceridemia and hyperamylasemia).
Preliminary data from a phase II, partially blinded (to the dose of the active agent), noninferiority trial of elvitegravir versus a comparator PI (CPI) have been presented.45 This dose-finding study randomized 278 treatment-experienced patients to receive 20, 50, or 125 mg of elvitegravir with 100 mg of ritonavir versus a comparator-boosted PI with OBT. Subjects were stratified by enfuvirtide use as well and were required to have a viral load of ≥1000 copies/mL and ≥1 protease resistance mutations on resistance testing. There were no CD4 cell count exclusions. The OBT was not allowed to contain NNRTIs in any of the trial’s arms and new PIs such as darunavir and tipranavir were initially not allowed to be included in the OBT of those receiving elvitegravir due to concerns about drug–drug interactions. An independent Data Safety Monitoring Board (DSMB), however, liberalized the original protocol after week 8 of the study to allow addition of darunavir or tipranavir to the OBT in the elvitegravir arms when data on potential drug–drug interactions suggested this was safe. Subjects enrolled were mostly male (86–99%) and Caucasian (66–86%) and had fairly advanced disease (mean CD4 count from 157 to 243 cells/mm3; mean HIV-1 RNA levels were 4.47-4.71 log10 copies/mL. Subjects were highly treatment-experienced with about half having GSS of zero for all NRTIs in the OBT. The median number of PI mutations in the group was ∼10 and most subjects were enfuvirtide-experienced (76–83%). Median numbers of ARVs in the OBT were three (including enfuvirtide). The OBT of subjects in the CPI commonly included new PIs (49% darunavir and 27% tipranavir). At week 16, only four patients had added a PI to their OBT but at week 24, this had risen substantially to 15% of those receiving elvitegravir. Analysis of preliminary data at week 8 demonstrated high rates of virologic failure in the elvitegravir 20 mg-arm and the DSMB advised investigators to stop enrollment into this arm of the trial. Subjects originally randomized to receive 20 mg of elvitegravir were offered open-label elvitegravir 125 mg with ritonavir 100 mg daily. Because the study had enrolled patients rapidly, only four subjects in the elvitegravir arms added darunavir or tipranavir before week 16. By week 24, 15% of subjects in the elvitegravir arms had added a PI to their OBT. The analysis of noninferiority was, therefore, conducted on the week-16 data (with the four patients receiving a PI prior to this point excluded from data analysis). The study’s primary end-point was the time-weighted average change in plasma HIV-1 RNA from baseline at week 24 (DAVG24). Although subjects in the 50 mg elvitegravir arm of the trial did not achieve statistically significant difference in DAVG24, those in the 125 mg group demonstrated a significant difference in virologic response compared to those in the CPI arm (−1.7 log10 copies/mL for elvitegravir 125 mg with ritonavir 100 mg versus 1.2 log10 copies/mL in the CPI group). There was a trend toward superior virologic outcomes with elvitegravir, with 76% of subjects in the elvitegravir 125 mg-arm achieving 2 log10 copies/mL, compared to 51% in the CPI arm. Virologic response was maximal at 2 weeks and tended to level off by week 16 (viral load decline of 2 log10 copies/mL in the 125 mg-arm at week 2, compared to 1.7 log10 copies/mL at week 16). Addition of elvitegravir to OBT with one active agent resulted in a more sustained virologic response at week 24 (−0.7 log10 copies/mL when 125 mg elvitegravir added to OBT with no active drugs compared to −2.1 log10 copies/mL arm in subjects with active NRTI or first use of enfuvirtide). Although not statistically significant, mean CD4 count increase was higher in the 125 mg elvitegravir arm than the CPI arm (61 cells/mm3 versus 28 cells/mm3). Adverse events were common in all arms of the study, as were grade 3 and 4 laboratory abnormalities (14–18% and 21–32% respectively), but there were no differences between any of the arms and rates of treatment discontinuation were rare (1–3%).
Elvitegravir is known to be metabolized both through CYP3A oxidative and glucuronidation pathways and to be a moderate inducer of CYP3A,44 so the potential for drug–drug interactions is a concern. Pharmacokinetic studies in small groups of healthy adults did not demonstrate clinically relevant interactions between elvitegravir and either emtricitabine/tenofovir (Truvada),46 zidovidine,47 abacavir, didanosine, darunavir or tipranavir.48
Nucleoside Reverse Transcriptase Inhibitors
Although still important in the management of HIV-infected persons, current NRTIs (and the related nucleotide reverse transcriptase inhibitor, tenofovir), have toxicities that often limit their effectiveness and have also proven vulnerable to the development of resistance, fueling demand for new agents in this class. NRTI development has focused on elucidating the mechanisms of drug toxicity and identifying agents that are less toxic and retain activity in the presence of known resistance mutations.
D-d4FC (Reverset, DPC 817, Dexelvucitabine)
D-d4FC is the D-enantiomer of the cytodine analog, dideoxyfluorocytidine. Although D-d4FC was withdrawn from development, experience with the drug is instructive for development of similar compounds. Animal studies of D-d4FCsuggested that it possessed good bioavailability and favorable pharmacokinetics.49 Similarly, early human studies indicated that the agent had oral bioavailability, reasonable pharmacokinetics and antiviral activity against many viruses resistant to other nucleosides.51–52 Clinical trials subsequently demonstrated substantial antiviral activity of D-d4FC in treatment-experienced subjects.53–55 However, in one of these trials, interesting interactions were observed between D-d4FC and other NRTIs.55 Viral load declines were much greater for subjects who were not receiving lamivudine or emtricitabine, suggesting a potential negative interaction between D-d4FC and other cytidine analogs. In vitro synergy studies had suggested that D-d4FC has additive or synergistic activity with NRTIs (zidovudine and stavudine), NNRTIs (nevirapine and efavirenz) and PIs (nelfinavir, lopinavir, and saquinavir).56 In addition to demonstrating reduced virologic efficacy, administration of D-d4FC with didanosine was associated with a dose-dependant increased risk of pancreatitis. Among subjects in the 200 mg D-d4FC daily group who were also receiving didanosine, seven of 14 developed grade IV hyperamylasemia and several were symptomatic. Pancreatitis tended to emerge several weeks into the study and all subjects recovered completely once the D-d4FC was stopped. Although the risk was amplified when it was co-administered with didanosine, the rates of Grade IV hyperamylasemia in subjects receivingD-d4FC were judged to be unacceptably high, and, thus, furtherdevelopment of this compound was discontinued.
Elvucitabine (ACH-126,433, β-L-d4FC)
Elvucitabine is another cytidine analog with activity against both HIV-1 and hepatitis B virus (HBV). Elvucitabine is the L-enantiomer of D-d4FC and has demonstrated similar anti-HIV-1 activity.57 In vitro studies suggest that elvucitabine is at least 10-fold more active than lamivudine and fivefold more active than emtricitabine.58 Initial reports also suggested a low risk for mitochondrial toxicity with elvucitabine monotherapy59 and in combination with other NRTIs.60 Studies of elvucitabine against both wild-type HIV-1 and clinical isolates with NRTI and NNRTI resistance demonstrated IC50 between 0.1 and 0.3 μM; these studies included viruses with M41L, T215Y, Q151M and the NNRTI resistance mutations K103N, Y181C, and G190A.61 Although isolates with the M184V mutation demonstrated reduced susceptibility to elvucitabine, IC50 only rose to 1–4 μM, an IC50 comparable to that of lamivudine against wild-type virus. Serial passage studies suggest that elvucitabine selects for mutations at position 184, though in an unusual way.62 Although the development of an M184V mutation was seen, investigators more typically observed the simultaneous emergence of M184I and D237E substitutions. An in vitro combination study suggested synergy between elvucitabine and either zidovudine or stavudine and an additive activity when combined with didanosine.63
A proof of concept, open-label study of the safety and efficacy of elvucitabine has been performed.64 Investigators recruited subjects on a failing ARV regimen with documented M184V mutations to assess the impact of substituting elvucitabine for lamivudine in their current three-drug regimen. Study subjects were randomized 25:25:10 to substitute 50 or 100 mg daily of elvucitabine for lamivudine for 28 days or to continue their baseline regimen (the control group). Investigators treated 56 subjects whose mean CD4 count was 471 cells/mm3 and whose mean plasma HIV-1 RNA level was 10 300 copies/mL. Elvucitabine-treated subjects showed a mean decline in HIV-1 load of 0.67 and 0.78 log10 copies/mL for the 50 and 100 mg groups, respectively, compared to a rise of 0.01 log10 copies/mL for the control group. No change was seen in CD4 cell count during the study period. Unfortunately, the study was discontinued prematurely when four individuals showed signs of bone marrow suppression (as evidenced by a decline in both total white blood cell count and absolute neutrophil count). Bone marrow toxicity emerged during weeks 3 or 4 of the study and was more common in subjects receiving 100 mg/day of elvucitabine. Extrapolating from data from HBV treatment trials that used lower doses of elvucitabine, pharmacokinetic and pharmacodynamic modeling suggested that reducing both dose and frequency of administration could reduce the risk of bone marrow toxicity while maintaining antiviral activity.65 This hypothesis has been tested in HIV-infected subjects.66 Investigators treated 24 HIV-infected individuals with either 5 or 10 mg daily or 20 mg every other day for21 days with lopinavir/ritonavir 400/100 mg twice daily. Plasma HIV RNA declined by 1.8 log10 copies/mL, 1.9 log10copies/mL, and 2.0 log10 copies/mL at day 21 in the 5 mg daily, 10 mg daily and 20 mg every other day groups, respectively. Concentrations of elvucitabine were still three times the IC50 for wild-type virus at day 28. No resistance to elvucitabine was demonstrated in this study, and no hematologic or other toxicity was reported. A follow-up 7-day monotherapy study randomized 24 subjects 1:1 to receive 10 mg of elvucitabine daily or placebo.67 Recognizing the long T½ of elvucitabine, this trial called for administration of lopinavir/ritonavir twice daily for 21 days after discontinuation of the study drug to avoid development of resistance. At day 7, mean HIV-1 RNA decline was 0.85 log10 copies/mL and this decline continued in a concentration-dependant manner after the study drug was stopped. Elvucitabine levels remained above the IC50 until day 21. Mean CD4 counts rose 62 cell/mm3 at day 7 in those receiving elvucitabine. There were no adverse events or significant laboratory abnormalities reported in this short trial and emergence of resistance was not observed. Phase II studies of elvucitabine versus lamivudine in combination with tenofovir and efavirenz are currently underway to assess the virologic and immunologic activity and safety of this compound in larger numbers of subjects.
Apricitabine (AVX-754, SPD-754, BCH-10618, (–)dOTC)
Apricitabine is the negative enantiomer of the cytidine analog racemate, dOTC and is structurally similar to lamivudine. In vitro studies of apricitabine against wild-type viruses demonstrated an IC50 between 0.02 and 4.2 μM.68 Apricitabine retained activity against a panel of HIV-1 isolates with up to 5 thymidine-associated mutations (TAMs).69 When tested against recombinant viruses with the M184V mutation, apricitabine’s activity declined by a median of 1.8-fold and was further reduced (approximately threefold) when viruses were engineered with both the M184V mutation and 3 TAMs. Serial passage studies demonstrated the emergence of K65R, V75I, and M184V mutations.68 K65R and V75I resulted in a 1.6- to 4.3-fold reduction in apricitabine activity. Although initial in vitro synergy studies of apricitabine suggested favorable interactions with lamivudine (as well as with stavudine, didanosine, zidovudine, and abacavir), subsequent studies of intracellular metabolism demonstrated a sixfold reduction in intracellular apricitabine triphosphate when administered with lamivudine.70 This finding was particularly surprising because co-administration of apricitabine with lamivudine resulted in nearly identical plasma pharmacokinetics. Studies of co-administration with emtricitabine demonstrated similar isolated declines in intracellular apricitabine triphosphate levels,71 and ruled out the possibility that this effect was produced by inhibition of apricitabine entry into cells.72
Safety and pharmacokinetics of a single dose of apricitabine were evaluated in a phase I trial in HIV-uninfected volunteers.73 Twenty-six subjects were divided into three cohorts and received 400, 800, or 1600 mg of apricitabine. Absorption was rapid with Tmax ranging from 1.5 to 1.7 h. Bioavailability was good and drug exposure increased with dose. Over 80% of apricitabine was eliminated unchanged in the urine, reducing concerns about interactions with ARV agents that are hepatically metabolized. Studies of intracellular pharmacokinetics of apricitabine triphosphate showed a T½ of 6–7 h in target cells, supporting twice-daily dosing.74 Absorption of apricitabine is not significantly affected by administration with food.75
A placebo controlled trial in treatment-naive; HIV-1 infected volunteers evaluated the pharmacokinetics, efficacy and safety of 10 days of apricitabine monotherapy.76 Investigators treated 63 subjects, all of whom had a CD4 count >250 cells/mm3 and plasma HIV-1 RNA level between 5000 and 100 000 copies/mL, and assigned them to receive daily doses of apricitabine ranging from 400 to 1200 mg. The groups receiving apricitabine experienced declines in HIV-1 RNA levels which ranged from 1.18 to1.65 log10 copies/mL, compared to ∼0.18 log10 copies/mL for those receiving placebo. All subjects were screened for NRTI mutations at study entry and again at the completion of the trial. Four subjects demonstrated NRTI resistance at baseline (though none had an M184V mutation). When compared to subjects with wild-type virus, subjects with NRTI resistance at baseline experienced similar declines in HIV-1 RNA.77 None of the treated subjects developed new NRTI mutations during the study. The pharmacokinetics of apricitabine were similar to those seen in uninfected volunteers.78 Using both plasma and intracellular pharmacokinetic data from the 10-day monotherapy trial, investigators developed a mathematical model to characterize the viral dynamics of apricitabine therapy.79 This model has been used to help investigators select 800 mg twice daily as the appropriate dose for phase II studies.
Despite encouraging data on its activity, enthusiasm for apricitabine has been tempered by the fact that development of its parent compound, dOTC, was halted because long-term administration produced a degenerative dermopathy in animals. A 52-week study of apricitabine in cynomolgus monkeys suggested that these changes were due to the (+)dOTC enantiomer.80 The exact mechanism of this toxicity is uncertain but is probably not related to mitochondrial toxicity, as dOTC has not been shown to interfere with mitochondrial DNA expression.81
Racivir (RCV; (±)-FTC; ((±)-β-2′,3′-dideoxy-5fluoro-3′-thiacytidine))
Racivir is a 50:50 racemic mixture of two β-nucleoside enantiomers.82 Both enantiomers of racivir are potent inhibitors of HIV reverse transcriptase with in vitro studies demonstrating EC90 of 0.04 μM and 0.6 μM for (−)-FTC and (+)-FTC respectively.83 Mechanistic studies have shown that the negative enantiomer of FTC is taken up more efficiently into target cells, triphosphorylated more rapidly and degraded less readily than the positive enantiomer,84 perhaps explaining the former’s greater potency. Despite the successful licensing of (−)-FTC (emtricitabine), several distinct properties have led its developers to believe racivir will have an important role in the management of HIV infection. In vitro selection studies have shown that the two enantiomers of racivir select for different RT mutations. (M184V for the negative enantiomer and Y215Y for the positive). Its developers suggest that these dual pathways of resistance will reduce the likelihood of the emergence of resistance and point to the fact that racivir has a higher in vitro barrier to resistance with racivir compared to either lamivudine or emtricitabine85 as grounds for moving forward with development of this compound.
Single- and multiple-dose studies of the pharmacokinetics and safety of racivir in healthy volunteers have been performed.86 In this study, 32 subjects were randomized 4:1 to receive single 50, 100, 200, or 400 mg doses of racivir or placebo. Cmax and AUC were dose proportional for all doses and exceeded EC90 for wild-type HIV. The median T½ for racivir ranged from 1.8 h at 50 mg to 3.4 h at 400 mg. No SAEs were observed and side effects were generally mild (headache most commonly). At 400 mg, the Cmax for racivir was greater than the EC50 for lamivudine-resistant HIV.
No studies of racivir monotherapy have been reported though the results of a 14-day, dose-escalation study of combination ART with racivir has been published.87 In this randomized, placebo-controlled study, 25 HIV-1-infected, ART-naive subjects were randomized to receive 200, 400, or 600 mg of racivir along with standard doses of efavirenz and stavudine. The placebo group received lamvudine 300 mg daily. Racivir was administered once daily at doses of 200, 400, or 600 mg. At the end of the study, mean decreases in plasma RNA were 1.92 log10 copies/mL, 2.03 log10 copies/mL, and 2.17 log10 copies/mL for the 200, 400, and 600 mg racivir groups compared to 2.25 log10 copies/mL for the lamivudine group. All four groups displayed persistent suppression of peripheral HIV-1 RNA until day 28 (mean viral load 2.02–2.43 log10 copies/mL below baseline) at which time they began to return toward baseline. The most common adverse event was dizziness, which was probably attributable to efavirenz.82
The virologic efficacy of racivir in treating persons with documented M184V mutation has been studied.83 Investigators randomized subjects receiving lamivudine-containing ART to substitute racivir or to continue with lamivudine for 28 days. Subjects were required to be on a stable ARV regimen for at least 60 days and to have a CD4 count of 50 cells/mm3 with an HIV-1 RNA level of 2000 copies log10 copies/mL and a documented M184V mutation. Subjects were enrolled 2:1 to receive an unspecified dose of racivir versus placebo. Mean plasma HIV-1 RNA level was 4.1 log10 copies/mL. Viral load measurements at 28 days demonstrated a decline of 0.4 log10 copies/mL in those receiving racivir versus a rise of 0.14 log10 copies/mL in those who stayed on lamivudine. Subset analysis showed that most of the beneficial effect of racivir was observed in subjects with an M184V mutation and <3 TAMs. Mean viral load decline in these subjects was 0.7 log10 copies/mL, and 28% achieved a viral load of <400 copies/mL at the end of the study. Further studies of racivir as second-line therapy for patients with the M184V mutation are planned.
Non-Nucleoside Reverse Transcriptase Inhibitors
Soon after their introduction in the mid-1990s, NNRTIs assumed an important role in ART and several landmark studies established efavirenz as a preferred agent for the initial treatment of HIV-1 infection. Unfortunately, NNRTIs have demonstrated a low genetic barrier to resistance, with the most common point mutations (K103N and Y181C) rendering all three currently approved agents inactive.88 As rates of resistance to NNRTIs in both chronically and recently infected persons increase,85 there is a need for compounds that retain activity against viruses resistant to current NNRTIs. Development of second generation NNRTIs has been slowed by difficulties finding agents that are potent, nontoxic and maintain activity in the presence of NNRTI mutations.
TMC 125 (Etravirine, R165335)
TMC 125 was derived from the tetrahydro-imidazo[4,5,1-jk][1,4]-benzodiazepin-2 (1H)-one and -thione (TIBO) group of compounds.9,89 TIBO derivatives were among the first NNRTIs identified. Crystalographic analyses led to the recognition that TIBO derivatives were a new class of ARV compounds, and have allowed investigators to rationally design a series of compounds that maintain antiviral potency while expanding the spectrum of activity against viruses with resistance to first-line agents.90 Etravirine, a derivative of diarylpyrimidine (DAPY), inhibits wild-type HIV-1 in vitro with an EC50 of 1.4 nM.91 In contrast to other NNRTIs, etravirine has shown in vitro activity against HIV-2 as well, although at micromolar, rather than nanomolar, concentrations. Etravirine is equipotent against both wild-type and many NNRTI-resistant variants and retained activity against 97% of 1081 clinical isolates with NNRTI-associated mutations. It is active against a variety of HIV-1 subtypes (group M subtypes A, B, C, D, F, and H) and retains partial activity against HIV-1 group O isolates with the NNRTI mutations A98G, V179D, and Y181C. The enhanced spectrum of activity of etravirine results from multifaceted binding that produces conformational and positional stability despite changes in HIV-1 RT.92
Etravirine has a higher genetic barrier to resistance in vitro than currently available NNRTIs and requires the accumulation of multiple mutations before its activity is substantially impaired.93 Selection experiments using increasing concentrations of etravirine resulted in the accumulation of known NNRTI-associated mutations such as L100I, Y181C, G190E M230L, and Y318F, as well as the novel mutations V179I and V179F. Etravirine is lipophilic and has limited oral absorption, but has been reformulated to improve oral bioavailability and reduce pill burden.94 As a result, future phase III studies of etravirine are being performed with 200 mg of the tablet form of etravirine, which is bioequivalent to the previous 800 mg formulation. The absorption of etravirine is enhanced threefold when taken with food.95 Pharmacokinetic studies of etravirine in ARV-naive, HIV-1-infected subjects demonstrated a Tmax of 3 h and Cmax of 93 ng/mL when dosed at 900 mg twicedaily.96 Steady-state concentrations were achieved within5 days, at which time Cmax averaged 418 ng/mL and Cmin 245 ng/mL.
In a randomized, double-blind, placebo-controlled, phase II trial, seventeen treatment-naive subjects were randomized 2:1 to 900 mg of etravirine twice daily or placebo for 7 days.96 At the end of the study, etravirine-treated subjects experienced a decrease of 1.99 log10 copies/mL in plasma viral load versus 0.06 log10 copies/mL in the placebo arm. A retrospective analysis demonstrated rates of decline in HIV-1 RNA with etravirine monotherapy that were similar to those seen with a five-drug, triple-class regimen.97 An open-label, proof-of-concept study of subjects with documented NNRTI resistance demonstrated antiviral activity of etravirine in treatment-experienced subjects.98 Investigators studied sixteen subjects who had a plasma HIV-1 RNA level of >2000 copies/mL while on an NNRTI-containing ARV regimen, and substituted 900 mg of etravirine twice daily for their current NNRTI (efavirenz or nevirapine). At the end of the 7 daystudy period, the mean decline in HIV-1 RNA was 0.89 log10 copies/mL. Pretreatment genotypic analysis showed an average of two different NNRTI mutations in study subjects (range 1-4). Phenotypic studies demonstrated high-level resistance to all approved NNRTIs, but preserved susceptibility to etravirine.
Recently the 24-week results of two identically designed ongoing Phase III trials of etravirine (DUET-1 and DUET-2) were reported.99,100 Both trials enrolled adults with virologic failure on stable antiretroviral therapy, NNRTI resistance, VL > 5000 copies/ml and 3 or more PI-associated mutations. Subjects were randomly assigned to receive 200 mg of etravirine or placebo twice daily; all subjects also received darunavir/ritonavir together with investigator-selected NRTI. Enfuvirtide use was optional. Plasma viral load < 50 copies/ml was the primary endpoint.
In DUET-1, 304 subjects were randomized to etravirine and 308 to placebo.99 At week 24, 170 (56%) in the etravirine group and 119 (39%) in the placebo group had reached the study endpoint (p = 0.005). Adverse events were similar in both groups, except that there were more rashes on etravirine (20% vs 10%) and more diarrhea on placebo (20% vs 12%). In DUET-2, 295 subjects were randomized to etravirine and 296 to placebo.100 At week 24, 183 (62%) in the etravirine group and 129 (44%) in the placebo had reached the study endpoint (p = 0.0003). Adverse events were comparable in the two groups. In an accompanying editorial, it was suggested that by a pooled analysis of both studies, it appeared that etravirine also reduced clinical progression,101 though this was an unplanned post-hoc analysis, utilizing data from both studies. Preliminary analyses of resistance data from DUET-1 and DUET-2 have identified 13 mutations associated with decreased virologic responses to etravirine; the concurrent presence of 3 or more mutations were necessary to substantially reduce drug efficacy.
The successes of these initial studies led the drug’s developers to design a series of larger phase II/III trials of etravirine. Interim safety and tolerability data have been reported on one such study.102 Investigators from the Etravirine-C203 trial studied individuals with a plasma HIV-1 RNA level of 1000 copies/mL, three-class experience and resistance testing that documented susceptibility to two approved ARV agents. At enrollment, subjects were randomized to receive 400, 800, or 1200 mg of etravirine twice daily versus placebo and OBT. The OBT averaged 3-4 additional drugs and was required to include compounds with anti-HIV activity identified on resistance testing. Use of enfuvirtide was allowed in the OBT. Adverse events were common in both groups (92% in pooled treatment groups versus 91% in the placebo arm). Diarrhea, headache and rash were seen most commonly, though the differences between the etravirine and placebo groups were not significant. The only SAEs reported in the treatment arms were three cases of pancreatitis (versus one in the control group), and one case of pancytopenia. Grade 3/4 laboratory abnormalities were common, but the rates were similar in subjects receiving etravirine versus placebo (35% vs 32%).
Data from a 48 week phase II placebo-controlled dose-finding trial on the efficacy and tolerability of etravirine in subjects with multidrug resistant HIV-1 infection have been reported.103,104 At enrollment, subjects were required to have a plasma HIV RNA level of ≥1000 copies/mL with documented NNRTI resistance and three primary PI mutations. Median baseline HIV-1 RNA was 4.7 log10 copies/mL and CD4 count was 100 cells/mm3.105 Baseline phenotypic testing showed a median fold change of 41.4 for efavirenz compared to 1.7 for etravirine. One hundred and ninety nine subjects were randomized 2:2:1 to receive either 400 or 800 mg of etravirine twice daily versus placebo, along with an optimized background regimen that could include enfuvirtide. Intention to treat analysis demonstrated HIV-1 RNA declines of 0.88 log10 copies/mL, 1.01 log10 copies/mL, and 0.14 log10 copies/mL in the 400 mg, 800 mg and placebo groups, respectively. Not surprisingly, response rates were better when etravirine was added to a regimen containing one or more additional agents to which the virus was sensitive. Viral load declines averaged 1.67 log10 copies/mL for subjects with no NNRTI mutations in the 800 mg etravirine arm compared to 0.54 log10 copies/mL for those with three NNRTI mutations.104,105 Subjects receiving 400 mg and 800 mg etravirine achieved an HIV-1 RNA level of <50 copies/mL in 23% and 22% of cases, respectively, compared to 0% in the placebo arm. Virologic failure was observed in 9% of subjects in the combined etravirine arms, compared to 78% of those in placebo arm. At week 48, mean CD4 count rose by 58 and 61 cells/mm3 respectively, in the 400 and 800 mg etravirine arms, compared to a rise of 13 cells/mm3 in the placebo arm. Analyses of safety and tolerability of etravirine are complicated by the high rates of treatment discontinuations in the placebo arm which resulted in much shorter duration of exposure to OBT in the placebo arm (48 weeks in the etravirine arms compared to 18 weeks in those receiving placebo). Adverse events were nearly universal, with 99% of subjects in the combined etravirine groups reporting some AE. Grade 3 AEs were seen in 39% of subjects receiving etravirine versus 23% in the control arm, but Grade 4 AEs were equally common between the groups. Grade 3 or 4 AEs observed in more than three subjects receiving etravirine included four cases of pneumonia, and three cases each of abdominal pain, rash, hypertriglyceridemia, and pancreatitis. Four subjects died during the study, three in the 400 mg etravirine arm and one in the placebo arm. Only one of these (cardiopulmonary failure and myocardial infarction) was felt to be potentially attributable to etravirine. Grade 3 or 4 laboratory abnormalities were seen in 39% of the combined etravirine subjects compared to 35% in the placebo arm. Models derived from 24-week data predicted reductions in rates of progression to AIDS/death of between 31% and 39% over 1 year in persons receiving 800 mg of etravirine twice daily.106
Not all studies of etravirine have produced overwhelmingly positive results, however. The drug developers sought to assess the efficacy of etravirine versus investigator-selected PIs in subjects with at least one NNRTI-associated mutation but no prior PI exposure.107 Fifty nine subjects were randomized to receive 800 mg of etravirine twice daily versus 57 who received a PI-based regimen (62% lopinavir/ritonavir and 32% atazanavir/ritonavir). NRTI backbone was selected based on the results of resistance testing. Both NRTI and NNRTI resistance was common in both arms (∼80% of participants with at least one NRTI mutation and ∼70% with two or more NNRTI mutations). Despite the high number of NNRTI mutations, however, median fold change in susceptibility to etravirine at entry was only 2.0 compared to 129.8 for efavirenz. Although viral load decline in both arms of the study were comparable at 4 weeks (1.3 log10 copies/mL for etravirine and 1.5 log10 copies/mL for those receiving a PI), those receiving etravirine experienced an early virologic rebound, and the study was shut down by the study’s independent DSMB. Analysis of 12 week data from the etravirine arm showed a sharp decline in efficacy of etravirine in subjects with >2 baseline TAMs and/or the M184V mutation. Viral load responses to etravirine ranged from a decline of 2 log10 copies/mL in those with no NRTI mutations to <0.5 log10 copies/mL in those with three or more. Grade 3 and 4 AEs were uncommon in this study and were comparable across both arms. Central nervous system effects were comparable, and gastrointestinal and lipid abnormalities were more common in those receiving a PI. Although the presence of multiple NNRTI mutations reduced the antiviral activity of etravirine, multivariate analysis did not suggest that presence of K103N or Y181C mutations predicted response.
Etravirine is an inducer of CYP3A4 and its AUC and Cmax decline significantly when administered with efavirenz, nevirapine, and full-dose ritonavir.108 Etravirine is metabolized in vitro by UDPGT and CYP2C.109 Interactions with PIs include increased etravirine levels when given with indinavir and atazanavir, and reduced etravirine levels when it is administered with full-dose ritonavir. This pharmacokinetic relationship is bi-directional and levels of both indinavir and saquinavir drop substantially when co-administered with etravirine, whereas fosamprenavir levels rise, making appropriate dose adjustment advisable. Studies in healthy volunteers demonstrated no interaction between etravirine and didanosine.110 Addition of 800 mg of etravirine twice daily to the regimens of HIV-1-infected subjects with undetectable viral loads on a stable combination ARV regimen containing saquinavir or lopinavir/ritonavir demonstrated only slightly lower Cmax and AUC levels.111 Cmin for lopinavir was unaffected, arguing that the clinical impact of etravirine on lopinavir/ritonavir will be minimal. While no pharmacokinetic interaction was observed in a small study of HIV-infected subjects receiving both etravirine and TMC 124/ritonavir,112 administration of etravirine with tipranavir/ritonavir in healthy subjects resulted in 24% decrease in AUC compared to when etravirine was administered alone.113 Pharmacokinetic studies did not show clinically significant changes in either etravirine levels or a variety of other medications including ranitidine, omeprazole, or rifabutin. Levels of sildenafil drop substantially when given with etravirine, as do levels of clarithromycin, so the drug’s developers suggest dose adjustment or alternative medications when these drugs are used.




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


