Chapter 22 Tenofovir
Tenofovir disoproxil fumarate (tenofovir DF) is the oral prodrug of tenofovir (9-[(R)-(2-phosphonomethoxy)propyl]adenine) (PMPA), a nucleotide (nucleoside monophosphate) analog with activity against retroviruses and hepadnaviruses. Tenofovir is an acyclic nucleoside phosphonate compound (Fig. 22-1) designed to circumvent the first phosphorylation step necessary for activation of nucleoside analogs, such as zidovudine, didanosine, stavudine, lamivudine, and abacavir.1 The antiviral activity of unphosphorylated nucleoside analogs may be impeded by their low affinity for cellular nucleoside kinases. Tenofovir differs from the ‘classic’ nucleoside analogs in that it is less dependent on intracellular enzymes for activation.2 Tenofovir DF (Viread) received FDA approval in the United States in 2001, and was approved by the European Medicines Evaluation Agency in 2002. Truvada is a fixed-dose combination of tenofovir DF and emtricitabine (FDA approved in 2004). Atripla is a fixed-dose combination of tenofovir DF, emtricitabine, and efavirenz (FDA approved July 2006).
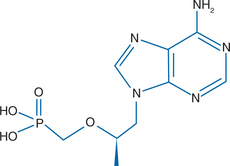
Figure 22-1 Structure of tenofovir disoproxil fumarate, or 9-[(R)-(2-phosphonylmethoxy)propyl]adenine.
MECHANISM OF ACTION AND IN VITRO ACTIVITY
Tenofovir DF is a lipophilic ester derivative of tenofovir designed to improve oral bioavailability. After oral administration and absorption tenofovir DF is rapidly cleaved by nonspecific extracellular carboxyesterases into tenofovir. Once inside cells tenofovir is metabolized by adenylate cyclase and nucleoside diphosphate kinase to tenofovir diphosphate (PMPApp), the active moiety. The antiviral effect of tenofovir is the result of selective interaction of PMPApp with the viral DNA polymerase or reverse transcriptase (RT). Based on the structural resemblance to natural deoxyadenine triphosphates (dATP), PMPApp acts as both a competitive inhibitor and an alternative substrate during the DNA polymerase chain reaction, resulting in DNA chain termination.1
Tenofovir has in vitro activity against hepadnaviruses (hepatitis B virus) and retroviruses.3–5 The spectrum of antiretroviral activity includes HIV-1 and HIV-2, simian immunodeficiency virus (SIV), feline immunodeficiency virus, visna-maedi virus of sheep, and murine leukemia and sarcoma viruses.6–9 In vitro activity against non-B HIV-1 subtypes has also been described. The mean 50% inhibitory concentration (IC50) values for tenofovir against HIV-1 subtypes A, C, D, E, F, G, and O in primary peripheral blood mononuclear cell (PBMC) cultures were all within twofold of the HIV subtype B IC50 value (range 0.55-0.22 μM).10
Tenofovir has demonstrated an anti-HIV effect in both lymphocytes and macrophages.11 The KI value of PMPApp against HIV-1 RT is 0.022 μM, which is ∼200-, 3700-, and 2700-fold lower than the KI value against human DNA polymerases α, β, and γ, respectively.12 Moreover, tenofovir and tenofovir DF were evaluated in vitro for antiviral activity (IC50) and cytotoxicity (CC50) using both laboratory and clinical HIV-1 strains; they were noted to have favorable selectivity indices (CC50/IC50 ratio higher than 2000).13 The antiviral activity of tenofovir against wild-type laboratory strains of HIV-1 ranges from 0.2 to 6.0 μM.13 In vitro, PMPApp (KI 0.022 μM) is slightly less potent than the active phosphorylated metabolite of zidovudine (KI 0.008 μM).12 The potency of tenofovir DF monotherapy (300 mg tablet daily) was assessed in 10 antiretroviral-naive HIV-infected individuals who underwent frequent monitoring of plasma HIV-1 RNA levels over 21 days. The individual first phase decay slopes of plasma HIV-1 RNA levels from −0.24 to −0.59 log10 copies/mL (median −0.40 log10 copies/mL) which is similar to the values previously described with ritonavir monotherapy indicating comparable potency between the two drugs. The mean reduction in viral load was 1.5 log10 copies/mL by day 21.14
In vitro analysis of MT2 cells infected with the HIV-IIIb strain were utilized to assess the activity of tenofovir in combination with a variety of other antiretroviral agents. The combination of tenofovir with zidovudine, amprenavir, and all non-nucleoside reverse transcriptase inhibitors (NNRTIs) tested demonstrated strong synergistic anti-HIV activity, whereas combinations with didanosine, adefovir, and nelfinavir resulted in minor synergistic inhibition of in vitro HIV replication.15 Additive HIV inhibition was noted with tenofovir in combination with abacavir, stavudine, lamivudine, zalcitabine, ritonavir, saquinavir, and indinavir.15,16 No tenofovir-containing combination demonstrated antagonism.
PHARMACOKINETICS
The formulation of tenofovir DF as an oral prodrug for tenofovir has allowed improved oral bioavailability. In vitro metabolic studies using radio-labeled tenofovir and tenofovir disoproxil fumarate in resting and activated peripheral blood lymphocytes demonstrated a more than 1000-fold higher intracellular concentration of the active PMPApp after incubation with the prodrug compared to tenofovir alone.13 Owing to its increased cellular permeability, the anti-HIV activity of tenofovir DF is increased by 17- to 90-fold over tenofovir. The diphosphorylation of tenofovir in cells to PMPApp is independent of viral infection, and there is evidence that these active intracellular metabolites persist in cells after the drug is removed, suggesting that an antiviral effect may continue in the absence of repeated tenofovir exposure. Moreover, the long intracellular half-life of PMPApp (10 to 50 h) allows once-daily dosing. Animal models have demonstrated that ∼98% of an intravenous tenofovir dose is eliminated unchanged in the urine after 24 h.13 Tenofovir is not a substrate, or inhibitor of the CYP450 enzyme system and does not have significant binding to plasma proteins.17
The pharmacokinetics of tenofovir and tenofovir DF has been studied in HIV-1-infected subjects. An aqueous solution of tenofovir was administered intravenously in single doses of 1 mg/kg and 3 mg/kg, resulting in mean peak serum concentrations of 2.7 and 9.1 μg/mL, respectively.18 Serum tenofovir concentrations declined in a biexponential manner with a terminal half-life of 7.1 h. All subjects in the higher dose (3 mg/kg) group had quantifiable serum levels up to 24 h after the initial dose. In a randomized, double-blind, placebo-controlled phase I/II study (GS-97-901) in HIV-infected individuals, the pharmacokinetics of tenofovir DF were studied at doses of 75, 150, 300, and 600 mg given in tablet form. For each dose cohort, fasting patients received a single oral dose; and after a 1-week observation period the same patients received single oral doses after ingesting a high-fat meal for 28 consecutive days. After fasting, the median peak plasma concentrations (Cmax) of tenofovir DF were 68.6, 111, 240, and 618 ng/mL in the 75, 150, 300, and 600 mg groups, respectively. Tenofovir has a long terminal half-life in serum of ∼17 h. At steady state (day 15), median Cmax values were −80.8, 163, 303, and 633 ng/mL and area under the serum concentration time curve (AUCss) values of 717, 1613, 2937, and 6073 ng h−1 mL21 in the same dosing groups when the drug was administered in a fed state.19 The oral bioavailability of tenofovir DF when administered with food was enhanced (∼40%) and was not affected by repeated dosing. In general, tenofovir DF displayed linear pharmacokinetics over the 75 to 300 mg/day dose range and was rapidly converted to the active form (tenofovir) following oral absorption with this process facilitated by administration in the fed state.19
In a recent study, single-dose and steady-state pharmacokinetics of tenofovir DF (300 mg daily) were evaluated in HIV-infected children and the AUC and Cmax were comparable to those values seen in HIV-infected adults.20
TOXICITY
The preclinical toxicology studies in animals (rats, dogs, monkeys) administered tenofovir over a minimum of 11 months identified the kidney and bone as potential target organs. Specifically, urinary excretion of calcium and phosphorus was increased in rats and dogs when tenofovir exposure was 11- and ninefold larger than standard human exposure, respectively; these findings were reversible and not seen when exposure was increased three- to fivefold over standard human exposure. Moreover, glucosuria and proteinuria were described in dogs and monkeys at nine- and 12-fold increased exposure but again were reversible and not present when dosing was three- to fourfold larger than standard human exposure. Decreased bone marrow density and increased parathyroid hormone secretion were noted in animals when dosing was increased nine- to 50-fold over standard human exposure. These bone toxicities were thought to be secondary to renal toxicity and were reversible with discontinuation of the drug or not seen at all when exposure was only two- to fourfold larger than standard human exposure. Other adverse effects noted in animal studies were gastritis, duodenitis, and slight elevations in liver transaminases; these findings were limited to rats in the higher dosing groups.21
Over 1600 HIV-infected individuals have received tenofovir DF alone or in combination with other antiretroviral agents in clinical trials, including a number of study participants who had been treated for 3 years or more. In general, tenofovir DF was well-tolerated. In a randomized, double-blind, placebo-controlled phase II study (GS-98-902), HIV-infected patients received tenofovir DF 75, 150, 300 mg or placebo once daily for 24 weeks in addition to the patient’s existing stable antiretroviral regimen at the time of enrollment. At week 24, placebo patients received tenofovir DF 300 mg once daily and all treatment groups were followed for a total of 48 weeks (see section on Efficacy). At week 24, the number of grade 3 or 4 adverse events seen in subjects receiving tenofovir DF was similar to those receiving placebo (14-19%), and the percentage of grade 3 or 4 laboratory abnormalities was also similar amongst the treatment and placebo groups.22 There was no evidence of dose-related increase in adverse events. In a separate randomized, double-blind, placebo-controlled phase III study (GS-99-907), HIV-infected patients received tenofovir DF 300 mg or placebo once daily for 24 weeks in addition to their existing antiretroviral regimen. At week 24, placebo recipients received tenofovir DF 300 mg once daily and were followed for a total 48 weeks (see section on Efficacy). As noted in the phase II study, the number of grade 3 or 4 adverse events and laboratory abnormalities were not substantially different between the placebo and treatment groups.23 A combined list of reported grade 3 or 4 laboratory abnormalities in these studies is presented in Table 22-1.
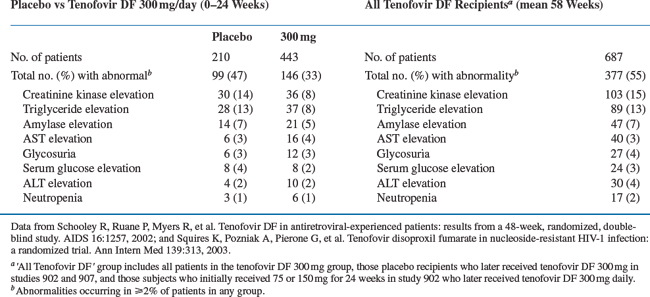
Interestingly, there have been no grades 3 or 4 serum creatinine elevations associated with tenofovir DF administration during phase II or phase III clinical trials.24 These findings are particularly significant given the high rate of proximal renal tubular dysfunction described with the structurally similar compound adefovir dipivoxil. The frequency of grade 3 or 4 serum hypophosphatemia in clinical trials was low (∼1%) and reversible with discontinuation of treatment.24 However, since completion of these clinical trials and with expanded use of tenofovir DF in clinical practice, there have been several small case series or case reports describing proximal renal tubular dysfunction and/or hypophosphatemia in HIV-infected patients suggesting longer follow-up may be necessary to fully assess renal toxicity.24–26 In these studies, proximal renal tubular dysfunction was diagnosed on average 7 months after initiation of tenofovir DF therapy and resolved on average 5 weeks after discontinuation of the drug.24 In a 3-year, randomized, multicenter, double-blind study (GS-99-903) involving 600 HIV-infected antiretroviral-naive individuals, the prevalence of grades 1, 2, and 3 serum creatinine elevations at week 144 were 4%, <1%, and 0%, respectively, in the tenofovir DF group and 2%, 0%, and <1% in the comparator (stavudine) group. Similarly, there were no differences in the prevalence of grade 1, 2, or 3 serum hypophosphatemia between the groups (4%, 3%, and <1% in the tenofovir group vs 4%, 2%, and <1% in the comparator group).27,28 Prior adefovir dipivoxil therapy does not appear to be associated with an increased risk of nephrotoxicity.
Bone mineral density testing was performed in a small proportion of subjects participating in the phase II study mentioned above (GS-98-902) and no clinically significant decrease in bone mineral density from baseline was demonstrated in patients receiving tenofovir DF through 48 weeks.22 Conversely, in a larger study with extended follow-up (GS-99-03, 144 weeks), a greater mean percentage decrease from baseline in bone mineral density at the lumbar spine was observed in participants receiving tenofovir DF when compared to those receiving stavudine (−2.2% vs −1.0%, respectively (P = 0.001)).28 In the same trial, bone mineral density changes in the hip were similar for both groups and the number of patients who experienced bone fractures during the trial was less in the tenofovir DF group.28
To date, adverse events associated with mitochondrial dysfunction have not been common. Lactic acidosis has occurred rarely in patients receiving tenofovir DF 300 mg daily in clinical trials and the majority of these subjects were receiving stavudine or didanosine concomitantly.29,30 These data correlate with the in vitro studies suggesting tenofovir DF is less toxic to mitochondria and has less effect on mitochondrial DNA polymerase than stavudine, zalcitabine, and didanosine.12,31
EFFICACY
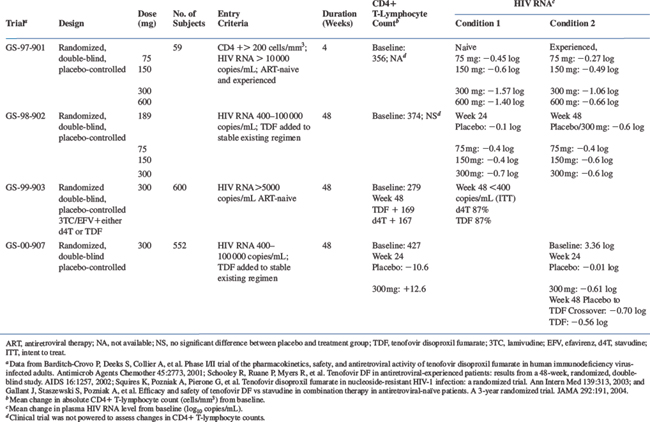
The clinical efficacy of tenofovir DF as measured by absolute CD4+ T-lymphocyte count or plasma HIV-1 RNA levels is shown in Table 22-2.
Tenofovir Disoproxil Fumarate Monotherapy
In a phase I/II randomized, double-blind, placebo-controlled, dose escalation study (GS-97-901), antiretroviral-naive and experienced adults with absolute CD4+ T-lymphocyte counts of 200 cells/mm3 or more and plasma HIV-1 RNA levels of 10 000 copies/mL or higher were administered tenofovir DF at once-daily doses of 75, 150, 300, or 600 mg or placebo for 4 weeks.19 Antiretroviral-naive patients receiving tenofovir DF had mean HIV-1 RNA level decreases of 0.45, 0.60, 1.57, and 1.40 log10 copies/mL in the 75, 150, 300, and 600 mg groups, respectively. Antiretroviral-experienced patients receiving tenofovir DF had mean HIV-1 RNA level decreases of 0.27, 0.49, 1.06, and 0.66 log10 copies/mL in the 75, 150, 300, and 600 mg groups, respectively. Overall, a dose-related treatment effect was observed, and individuals receiving 300 or 600 mg once daily had an overall mean HIV-1 RNA level reduction of 1.20 and 0.84 log10 copies/mL, respectively, compared to baseline, whereas placebo recipients experienced a 0.03 log10 copies/mL increase over the same period.
Combination Therapy with Tenofovir Disoproxil Fumarate
In a randomized, double-blind, placebo-controlled phase II study (GS-98-902), 189 HIV-infected patients received tenofovir DF (75, 150, 300 mg) or placebo once daily for 24 weeks in addition to the patient’s existing stable antiretroviral regimen (four or fewer drugs) at the time of enrollment. At week 24, placebo patients rolled over in a blinded fashion to tenofovir DF 300 mg once daily, and all treatment groups were followed for a total of 48 weeks.22,32 Mean baseline plasma HIV RNA levels were 3.8, 3.6, 3.6, and 3.7 log10 copies/mL in the placebo and the 75, 150, and 300 mg treatment groups, respectively. Study participants were highly treatment-experienced with a mean of 4.6 years on antiretroviral therapy and a baseline mean absolute CD4+ T-lymphocyte count of 374 cells/mm3. In addition, baseline genotyping demonstrated that 94% of patients had nucleoside RT-associated mutations, 57% had protease inhibitor (PI)-associated resistant mutations, and 32% had non-nucleoside RT-associated mutations.
Through week 24, the mean change in plasma HIV RNA levels from baseline were −0.1, −0.4, −0.4, and −0.7 log10 copies/mL in the placebo and 75, 150, and 300 mg groups, respectively. Through week 48, the mean changes in HIV RNA levels from baseline were −0.4, −0.6, and −0.6 log10 copies/mL in the 75, 150, and 300 mg groups, respectively. Placebo recipients were administered tenofovir DF (300 mg) at week 24 resulting in a mean plasma HIV RNA level reduction from baseline of 0.6 log10 copies/mL through week 48.22,32
In a randomized, double-blinded, placebo-controlled phase III study (GS-99-903), 600 antiretroviral subjects were randomized to efavirenz (an NNRTI) and lamivudine with either stavudine (40 mg twice daily)/tenofovir DF placebo or tenofovir DF (300 mg once daily)/stavudine placebo.28 The participants’ baseline plasma HIV RNA levels and absolute CD4+ T-lymphocyte counts were 81 300 copies/mL and 279 cells/μL, respectively. At week 48, the intent-to-treat (ITT) analysis demonstrated equivalence with 87% of subjects in each group achieving plasma HIV RNA levels <400 copies/mL. Similarly, 82% and 81% of subjects achieved plasma HIV RNA levels <50 copies/mL in the tenofovir DF and stavudine groups, respectively. The absolute CD4+ T-lymphocyte count increased by 169 cells/μL in the tenofovir DF group compared to a 167 cells/μL increase in the stavudine group.28
In a separate randomized, double-blind, placebo-controlled phase III study (GS-00-907), 550 antiretroviral-experienced HIV-infected patients received tenofovir DF 300 mg once daily or placebo for 24 weeks in addition to their existing antiretroviral regimen. After week 24, placebo recipients were allowed to receive tenofovir DF for the remainder of the 48-week study period. At baseline, patients had a mean plasma HIV RNA level of 3.36 log, a mean absolute CD4+ T-lymphocyte count of 427 cells/mm3, and had received antiretroviral therapy for a mean 5.4 years. Baseline genotypic analyses in a subset of patients (n = 253) demonstrated that 94% of participants had nucleoside RT-associated mutations, 58% had PI-associated resistant mutations, and 48% had non-nucleoside-associated RT mutations.23
Through week 24, the mean change in plasma HIV RNA levels from baseline were −0.61 and −0.03 log in the tenofovir DF and placebo groups, respectively (P < 0.001). In addition, 45% of tenofovir DF recipients achieved plasma HIV RNA levels <400 copies/mL compared to 13% in the placebo group (P < 0.001). Reduction in plasma HIV RNA levels to <50 copies/mL was seen in 22% of those receiving tenofovir DF compared to 1% of placebo recipients (P < 0.001). The mean change in absolute CD4+ T-lymphocyte counts from baseline were +12.6 and −10.6 cells/mm3 in the tenofovir DF and placebo groups, respectively (P = 0.008). Through week 48, the prevalence of tenofovir DF recipients with plasma HIV RNA levels <400 and <50 copies/mL was 41% and 18%, respectively, suggesting a sustained response.23
The role of triple nucleoside analog RT inhibitor regimens for the treatment of HIV-1infection has been investigated. The once-daily triple nucleoside/nucleotide analog combination of abacavir (600 mg), lamivudine (300 mg), and tenofovir DF (300 mg) appeared advantageous relative to potency and tolerability, but was associated with an unacceptably high rate of virologic failure based on preliminary data from two clinical trials which were subsequently discontinued.33,34 Similarly, a 24-week pilot study evaluating the efficacy of once-daily didanosine EC (250 mg), lamivudine (300 mg), and tenofovir DF (300 mg) in antiretroviral-naive HIV-infected individuals demonstrated suboptimal virologic responses. At week 12, only one participant demonstrated an optimal response (defined as = 2 log reduction in plasma HIV-1 RNA level); the median change from baseline plasma HIV-1 RNA level was −0.61 log.35 The reason for the poor performance of these regimens appears to be due to the early development of resistance, particularly the presence of the K65R and M184V mutations.33
In an effort to assess the efficacy of a once-daily regimen, an open-label, prospective, randomized, noninferiority study was performed in 517 HIV-1-infected, antiretroviral-naive patients comparing once-daily tenofovir DF (300 mg), emtricitabine (200 mg), and efavirenz (600 mg) to a fixed dose of zidovudine (300 mg) and lamivudine (150 mg) twice daily (Combivir) and efavirenz (600 mg) daily. Through week 48, significantly more patients in the tenofovir–emtricitabine group reached and maintained the primary end point (plasma HIV RNA of <400 copies/mL) than did those in the zidovudine–lamivudine group (84% vs 73%).36 Significant differences were also seen in the proportion of patients with plasma HIV RNA levels <50 copies/mL (80% in the tenofovir–emtricitabine group vs 70% in the zidovudine–lamivudine group) and in increases in absolute CD4+ T-lymphocyte counts (190 vs 158 cells/mm3, respectively).36

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


