Chapter 11 Abacavir
STRUCTURE
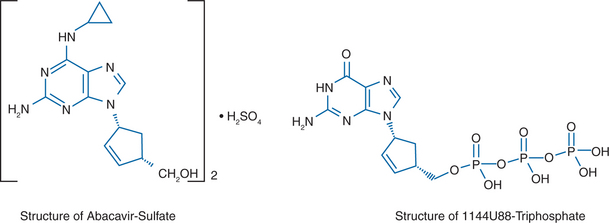
Abacavir sulfate, compound 1592U89 hemisulfate ((1S, cis)-4-(2-amino-6-(cyclopropylamino)-9H-purin-9-yl)-2-cyclopentene-1-methanol sulfate (salt)(2:1)) (formerly known as 1592), also known as Ziagen or as ABC, is a carbocyclic nucleoside analog with potent and selective activity against HIV-1.1–8 This compound contains a novel 6-cyclopropylamino-substituted purine. The structure is shown in Figure 11-1A. The compound is activated by cellular enzymes to the triphosphate derivative of the guanine analog 1144U88 ((1R,4S)-9-(4-(hydroxymethyl)-2-cyclopenten-1-yl)-guanine; (−)-carbovir), which is shown in Figure 11-1B.
IN VITRO ACTIVITY AND MECHANISMS OF ACTION
The active form of abacavir, 1144U88 triphosphate, is a potent inhibitor of HIV-1 reverse transcriptase (RT) in vitro, with a KI of 0.02 μM.1–7 In whole virus assays, abacavir has activity against HIV-1 (strain IIIB) cultured in MT-4 cells (a human leukemic cell line transformed with HTLV-1), peripheral blood mononuclear cells (PBMCs), and macrophages. The 50% inhibitory concentrations (IC50s) in these cells were, respectively, 4.0, 3.7, and 0.65 μM.5 The mean IC50 for abacavir against eight fresh clinical isolates of HIV-1, obtained from zidovudine-naive patients and amplified in PBMCs, was 0.26 μM (i.e., equivalent in potency to zidovudine).1,3,5,7 Abacavir has also demonstrated synergistic activity in vitro against HIV-1 when used in combination with zidovudine, other nucleoside reverse transcriptase inhibitors (NRTIs) such as lamivudine and didanosine (but additive with stavudine) and non-nucleoside reverse transcriptase inhibitors (NNRTIs) such as nevirapine, and protease inhibitors (PIs) such as amprenavir.1,5,7–9 There was no evidence for an antagonistic interaction between tenofovir and abacavir, in the presence or absence of 3TC, against wild-type and drug-resistant HIV-1 in vitro, to account for the high rate of virologic failures seen in patients initiating therapy with combined tenofovir, abacavir, and lamivudine.10
PHARMACOKINETICS
The cyclopropylamino moiety of abacavir is important for enhanced absorption and central nervous system penetration in vivo when compared to carbovir.5,11 In contrast to the limited oral absorption of carbovir (e.g., 26% and 23% oral bioavailability in the rat and monkey, respectively), pharmacokinetic evaluation of abacavir showed good oral bioavailability (e.g., 92% and 77% in mice and monkeys, respectively).5,12–14 In contrast to the poor brain penetration of carbovir (e.g., rat brain/plasma concentration ratios averaged 0.04), the penetration of abacavir into monkey cerebrospinal fluid (CSF) and rat brain were comparable to that of zidovudine.5 Abacavir showed significant penetration into CSF in humans.15 One observational cohort study (the Charter trial) in 374 highly active antiretroviral therapy (HAART)-experienced subjects with plasma and CSF viral load measurements reported a projected CSF penetration score (based on published data on CSF concentrations and/or chemical properties) that was highest for abacavir and zidovudine when compared to all other available NRTIs (including tenofovir, didanosine, zalcitabine, stavudine, lamivudine, and emtricitabine).16
Intracellular activation of abacavir to its active triphosphate form requires phosphorylation by cellular enzymes.2,4,6,17 This involves a unique set of enzymes other than those involved in activation of other NRTIs that are currently approved for treatment of HIV-1 infection. Abacavir sulfate is activated intracellularly to carbovir monophosphate by a novel phosphorylation pathway2–6 involving adenosine phosphotransferase.18 Carbovir monophosphate is then efficiently anabolized by cellular enzymes to carbovir diphosphate, followed by anabolism to the active form carbovir triphosphate, which inhibits HIV-1 RT. The existence of this unique activation pathway enables abacavir to overcome the deficiencies of carbovir (which include low oral bioavailability and minimal brain penetration) while maintaining potent and selective anti-HIV-1 activity.5,6 The intracellular half-life of carbovir triphosphate, produced from both abacavir and carbovir, is ∼3.3 h based on previous in vitro studies.2,5,19 However, more recent studies in vivo demonstrate that the intracellular half-life of carbovir triphosphate is much longer (20.64 h), which is possibly due to a saturation step and/or pooling of precursors (carbovir-monophosphate or carbovir-diphosphate).20 The prolonged half-life of carbovir-triphosphate observed in vivo supports the administration of abacavir once daily.20
The main route of elimination of abacavir is metabolism, with <2% of a dose recovered as unchanged drug in urine.15 Prolonged therapy of treatment-naive HIV-1-infected subjects over 48 weeks with combined regimens of efavirenz with either abacavir plus lamivudine, trizivir (a fixed-dose combination of Ziagen (abacavir), Retrovir (zidovudine), and Epivir (lamivudine)), or Combivir did not exhibit clinically significant changes in renal function as measured by glomerularfiltration rate.21 The catabolism of abacavir involves two pathways: glucuronidation, and carboxylation through alcohol dehydrogenase.13,14 The metabolism of abacavir is not dependent on cytochrome P450 liver enzymes. Physiologic concentrations of human albumin or α1-acid glycoprotein do not markedly alter the anti-HIV-1 activity of abacavir,22 and food intake does not significantly affect abacavir bioavailability. In an initial phase I double-blind, parallel, dose-escalation trial evaluating safety and kinetics, the tablet area under the concentration time curve (AUC) at the 300 mg dose was 99% of the oral solution AUC, and administration with food lowered AUC by 5% and maximum plasma concentration by 35%.23 The main route of metabolite excretion is renal, with 83% of a dose recovered in urine.15
There are no known significant drug interactions.24,25 Abacavir demonstrated predictable pharmacokinetic characteristics when administered as single oral doses ranging from 100 to 1200 mg.26 No significant pharmacokinetic interactions were detected after single-dose administration of abacavir in double or triple combinations with zidovudine, lamivudine, or both.27 In multiple-dose studies, abacavir showed predictable pharmacokinetic characteristics and zidovudine co-administration had no effect on the abacavir AUCtau (a parameter most closely associated with efficacy).28 In a prospective evaluation of intracellular concentrations and pharmacokinetics of carbovir triphosphate, tenofovir diphosphate, and lamivudine triphosphate in patients receiving the triple-nucleoside regimen of tenofovir disoproxil fumarate, abacavir, and lamivudine, there was no intracellular drug interaction that could explain the suboptimal viral response seen with this regimen.29 Changes in levels of dNTPs, carbovir-triphosphate, and tenofovir-diphosphate in purified human CD4+ and CD8+ peripheral blood T cells following treatment with tenofovir and/or abacavir and/or lamivudine in vitro similarly did not suggest that antagonistic drug–drug interactions occurred as the mechanism of regimen failure in vivo.30
In subjects with mild liver impairment (CNAB1006 Study), there was a 1.9-fold increase in abacavir exposure and a 1.6-fold increase in abacavir half-life in the group with liver disease. The liver disease did not modify the extent of formation of the metabolites, although the rates of formation and elimination were decreased. The CNAB1006 Study results suggested that patients with mild hepatic impairment should receive 150 mg abacavir twice daily in order to achieve an AUC equivalent to patients without liver disease receiving the recommended 300 mg twice-daily dose.31
The safety of abacavir and lamivudine-based HAART in 1985 treatment-naive HIV-1-infected subjects with and without hepatitis B and/or hepatitis C co-infection was examined using data from four large, randomized clinical treatment trials of abacavir plus lamivudine once daily or twice daily in combination with efavirenz or PIs. This 48-week analysis showed that these regimens were well tolerated in patients with hepatitis B and/or hepatitis C co-infection, with a similar median reduction in AST/ALT levels and a similar rate of grade 2–4 adverse events and drug-related adverse events when compared to non-co-infected subjects. Grade 2–4 increases in ALT and AST levels were more common in co-infected subjects (10.5% and 8.5%, respectively) when compared to non-co-infected subjects (1.3% and 1.3%, respectively), which was attributed to the natural history of viral hepatitis infection or to immune reconstitution leading to HBV immune-mediated response with ALT flares.32
Pharmacokinetics in HIV-1-Infected Infants and Children
A phase-I single-dose pharmacokinetic study evaluated two oral abacavir doses (4 and 8 mg/kg of body weight) in 22 HIV-1-infected children ages 3 months to 13 years.33 Abacavir was rapidly absorbed, with time to peak concentration in plasma occurring within 1.5 h postdosing. Abacavir was rapidly eliminated, with a mean elimination half-life of 0.98–1.13 h. The extent of exposure to abacavir appears to be slightly lower in children than in adults, with the comparable unit doses being based on body weight.33
TOXICITY
In Vitro Effects
Abacavir and carbovir are less myelotoxic than either zidovudine, didanosine, or zalcitabine when tested in vitro using human and murine hematopoietic progenitor cells.2,5 Carbovir triphosphate has relatively low inhibitory activity against cellular DNA polymerase γ when compared to the active triphosphates of other dideoxynucleoside analogs.5 It is the inhibition of cellular DNA polymerase γ that has been associated with drug-induced peripheral neuropathy and mitochondrial toxicity (including lipoatrophy/lipodystrophy). Taken together, the preclinical development of abacavir sulfate was supported by both a relative lack of myelosuppressive effects and less inhibition of cellular enzymes involved in development of peripheral neuropathy and other mitochondrial toxicities.
In Vivo Effects
In clinical trials to date, the majority of adverse events were rated as mild or moderate. No specific hepatic, pancreatic, renal, or bone marrow toxicity patterns have been described. In ACTG 5095, adding abacavir to a backbone regimen of combined zidovudine plus lamivudine plus efavirenz had no consistent metabolic effects in HIV-1-infected adults as measured by fasting triglyceride and total, LDL, and HDL cholesterol, lactate, glucose, and insulin resistance measurements.34 Mitochondrial toxicity (including lipoatrophy) occurring in association with treatment with abacavir may be lower than the risk with many other NRTIs. However, lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs alone or in combination, including abacavir, zidovudine, lamivudine, and other antiretrovirals. In a Commmunity Program for Clinical Research on AIDS (CPCRA) study, 96 antiretroviral-naive subjects were enrolled and randomized to didanosine plus stavudine (n = 46) or abacavir plus lamivudine (n = 50).35 Those subjects receiving didanosine plus stavudine experienced a decline in regional fat and total body fat when compared to increases in these parameters in subjects in the abacavir plus lamivudine arm (P < 0.05). Abacavir plus lamivudine therapy increased the rate of change of high-density lipoprotein cholesterol, whereas this rate decreased during didanosine plus stavudine therapy. Early and sustained increases in insulin resistance were seen only during didanosine plus stavudine therapy.35 In lipoatrophic HIV-1-infected adults enrolled in the RAVE study, switching from a thymidine analog to abacavir or tenofovir for 48 weeks improved lipoatrophy.36 Replacing the PI component of treatment regimens improved the lipid profile in 90 treatment-experienced subjects with controlled plasma viremia who were receiving nevirapine (n = 29), efavirenz (n = 32), or abacavir (n = 29) in the 24-month Spanish NEFA study, but did not seem to be effective for reversing body fat abnormalities.37 After 1 year, a trend toward a lower incidence of adverse effects leading to discontinuation among those subjects receiving abacavir was seen, but also a trend toward a higher rate of virological failure was noted.
A separate 48-week, randomized, open-label study of HIV-1-infected subjects compared three abacavir-base substitution approaches in the management of dyslipidemia and peripheral lipoatrophy.38 Subjects were receiving initial stavudine with either a PI or NNRTI and had hypercholesterolemia and/or lipoatrophy with well-controlled plasma viremia (plasma HIV-1 RNA ≤50 copies/mL). Thirty subjects were randomized to replace: (1) stavudine with abacavir; (2) PI or NNRTI with abacavir; or (3) stavudine and PI or NNRTI with abacavir plus zidovudine. Replacement of stavudine with abacavir led to modest improvements in fat mass in this study, and replacement of a PI or NNRTI with abacavir led to modest improvements in both cholesterol and triglycerides.
Abacavir Hypersensitivity Syndrome
Hypersensitivity reactions occurred in ∼8% of 2670 patients (n = 206) receiving abacavir for the treatment of HIV-1 infection in nine clinical trials (range: 2–9%) enrolled from Nov 1999 to Feb 2002 (Abacavir FDA prescribing information). The appearance of this syndrome demands immediate discontinuation and an absolute contraindication to reinitiation of abacavir or an abacavir-containing medication (e.g., Trizivir or Epzicom). A total of 1015 cases have been described among 26 769 subjects enrolled in phase II and III clinical trials and expanded access programs.39 The median time to onset was 11 days, with 93% of cases appearing within 6 weeks of initiation of abacavir, although reactions may occur at any time during therapy. In a risk factor meta-analysis for hypersensitivity reactions to abacavir introduction, the incidence observed was 3.7%. The odds ratios (OR) for subjects with previous HAART treatment and those of African descent were significantly lower (OR 0.41, 95% confidence intervals (CIs): 0.23, 0.72; and OR 0.51, 95% CI: 0.28, 0.92, respectively) than HAART-naive patients and other ethnic origins.40
The mechanism of abacavir hypersensitivity syndrome is not fully understood, but may involve an immunologic response. GlaxoSmithKline is conducting pharmacogenetic research to determine if cellular genetic DNA polymorphisms (variations) can be identified in HIV-1-infected patients who have developed hypersensitivity reactions following treatment with abacavir, in order to help predict risk of abacavir hypersensitivity in susceptible patients.41 An association between HLA-B*5701, HLA-DR7, and HLA-DQ3 and the hypersensitivity syndrome has been reported.42,43 A strong association between this syndrome and HLA-B*5701 carriage in Caucasians has been confirmed.44 Conversely, a lower reported rate of suspected abacavir hypersensitivity reactions among African-American patients across five randomized controlled trials has been attributed to a lower prevalence of HLA-B*5701 among African-Americans.45 A cutaneous patch test may help identify subjects with true abacavir hypersensitivity syndrome, since the syndrome may be difficult for clinicians to recognize.46 HLA-B*5701 was present in all seven patch test-positive subjects with remote abacavir hypersensitivity syndrome versus one of the 11 controls tolerating abacavir (P < 0.001). Five of seven patch tests (71%) versus one of 11 controls (9%) (P = 0.005) showed significant abacavir-specific CD8+ T-cell proliferation, suggesting a direct role for HLA-B*5701-restricted CD8+ T cells in the pathophysiology of abacavir hypersensitivity syndrome.47
The usual presentation includes symptoms indicating involvement of multiple organ systems.48 The most common symptoms are fever, rash, GI symptoms (nausea, vomiting, diarrhea, or abdominal pain), and malaise or fatigue. Fever is a key feature of the syndrome. Less common symptoms include respiratory symptoms (cough, sore throat, or dyspnea) and musculoskeletal complaints (myalgia, myolysis, arthralgia, edema) and headache and paresthesia. Hypotension may be present in 5% of reactions. In contrast to immunoglobulin E (IgE)-mediated anaphylactic reactions, wheezing is distinctly uncommon, and edema less common. In contrast to described reactions to NNRTIs, the rash is rarely severe, usually appearing later (e.g., 1–3 days) after the onset of the systemic symptoms, and may be absent in up to 30% of cases.49,50 The rash is usually maculopapular or urticarial. Morbidity is related more to the systemic symptoms rather than the rash. Although the time course is variable, symptoms tend to occur in succession over the course of several days, with fever and GI symptoms appearing early and rash later. The severity of symptoms tends to increase with each dose. A delay in discontinuation of abacavir in the face of an active hypersensitivity reaction may lead to a severe reaction and even death, although the risk of a fatal reaction is far more likely in the setting of a rechallenge (see further ahead).
After discontinuation of abacavir, symptoms improve quickly and resolve over a few days, although the rash, if present, may persist longer. If abacavir is reinitiated (rechallenge), symptoms reappear within hours and may be more severe. Fever, rash, malaise, hypotension, and facial and/or throat swelling or bronchospasm have been described. Symptoms not present with the initial reaction may also appear on rechallenge and temperature elevations to 39–40°C may occur. One case report described severe anaphylactic shock after rechallenge with abacavir without preceding hypersensitivity.51 Hypotension was present in ∼25% of cases where abacavir was reinitiated after an initial reaction. Support with intravenous fluids with or without vasopressors (dopamine/dobutamine) may be necessary. Fatalities have been reported.52 Among the reported cases where a rechallenge has been attempted, 4% of patients have died. The rechallenge reaction has occurred even at reduced doses of 100 mg abacavir. Thus, once hypersensitivity to abacavir has been diagnosed, do not rechallenge with abacavir or an abacavir-containing medication (e.g., Trizivir or Epzicom).
Diagnosis of ‘hypersensitivity reaction to abacavir’ occurred in 2–3% of subjects assigned to the nonabacavir-treated subjects in double-blinded studies.53 Most cases had rash alone or rash accompanied by nonspecific, mild symptoms, whereas hypersensitivity reactions to abacavir are part of a multisymptom syndrome with fever as the hallmark. Cutaneous reactions are common with other medications often prescribed to patients with HIV-1 infection, such as NNRTIs or sulfamethoxazole. The appearance or characteristics of a rash without other symptoms are not sufficiently specific to differentiate the cause in most cases. However, the morbidity associated with hypersensitivity reactions to abacavir is not related to rash, and rash is not frequently an early sign of this reaction. Thus, for patients in whom rash is the only clinical symptom, abacavir may be continued with the warning to follow the patient carefully for the development of other symptoms. The patient should be counseled to report the appearance of any additional symptoms immediately. In cases where systemic symptoms and rash are present together, differentiation may not be possible and abacavir should be discontinued. The rash of abacavir hypersensitivity is mild, but may involve mucous membranes. However, the Stevens–Johnson syndrome has only been reported when abacavir has been combined with NNRTIs.
In some cases, the symptoms of a hypersensitivity reaction may resemble acute infections or reactions to other medications. Keiser et al described the clinical features of diagnosed influenza infection with cases of hypersensitivity to abacavir.54 Both rash and GI symptoms were strongly associated with the latter. Respiratory symptoms with hypersensitivity to abacavir appeared in conjunction with GI symptoms. Nevertheless, for some cases differentiation of hypersensitivity to abacavir from an acute infection can be difficult. The symptoms will increase in severity if dosing is continued. Moreover, the timing of the onset of symptoms with respect to drug exposure is critical for recognition of the syndrome. Abacavir hypersensitivity starts several days to 6 weeks after starting the drug in most cases. Furthermore, the symptoms improve after stopping abacavir. Severe reaction and fatalities have occurred when symptoms of hypersensitivity to abacavir were ascribed to another cause and the discontinuation of abacavir was delayed or rechallenge was attempted. It is important not to ascribe the symptoms of abacavir hypersensitivity to nonspecific ‘flu’ or ‘gastroenteritis’.49,50
There is no evidence to indicate that interruption of abacavir administration in the absence of any symptoms (i.e., for a reason other than a side effect) increases the frequency or severity of a subsequent hypersensitivity reaction.55,56 However, there have been cases of hypersensitivity reactions occurring within 1 day of restarting abacavir where no prior symptoms of an initial hypersensitivity reaction were recorded. This underscores the importance to advise a patient to contact the healthcare provider if abacavir is discontinued. The healthcare provider should then obtain a careful history to assure that symptoms indicating a hypersensitivity reaction were not present at the time of interruption.
Most Common Adverse Events in Adult HIV-1-Infected Subjects Enrolled in CNAA2001
A summary of clinical toxicities associated with abacavir in the CNAA2001 trial is shown in Table 11-1.57,58 In that study, abacavir was administered as monotherapy for 4 weeks followed by randomization to therapy for 4 weeks followed in turn by randomization to zidovudine or its matching placebo for an additional 8 weeks. The most commonly reported adverse events were nausea, headache, asthenia, diarrhea, insomnia, fever, dizziness, vomiting, abdominal pain, and rash. Nausea was common, occurring in nine of 20 subjects (45%) at 300 mg PO bid. Headache occurred in eight of 20 subjects (40%) at 300 mg PO bid. A possible trend of increase in incidence of nausea and headache with increasing abacavir dose was observed. However, the small numbers of patients and the design of the study make it difficult to draw definitive conclusions about the significance of this preliminary observation. Only eight subjects (10%) withdrew from the study because of adverse events, all of which were considered possibly related to the study drug. The most common reasons for discontinuation were nausea (with or without vomiting) or hypersensitivity reaction. The latter occurred within 4 weeks of dosing. Only four subjects experienced a serious adverse event, none of which were considered related to the study drug.
Table 11-1 Most Common Adverse Events Associated with Abacavir in the CNAA2001 Triala
| Toxicity | Frequency | Comment |
|---|---|---|
| Nausea | 9/20 (45%) | A possible trend with increasing doses was noted |
| Headache | 8/20 (40%) | A possible trend with increasing doses was noted |
| Asthenia | 8/20 (40%) | |
| Diarrhea | 5/20 (25%) | |
| Insomnia | 5/20 (25%) | |
| Fever | 4/20 (20%) | |
| Dizziness | 2/20 (10%) | |
| Vomiting | 3/20 (15%) | |
| Abdominal pain | 3/20 (15%) | |
| Rash | ½0 (5%) |
a Adverse events experienced by the 300 mg bid cohort (n= 20 subjects) in the CNAA2001 trial, a phase I/II trial to evaluate the effects of multiple dosing of abacavir alone and in combination with zidovudine capsules. The majority of adverse events were rated as mild or moderate. The number of subjects listed experienced an event (regardless of association with abacavir) (see text).57 This table does not specifically list the occurrence rate of the abacavir hypersensitivity reaction, which has been estimated at 3% (range 2–5%) in abacavir-treated subjects.
Abacavir did not demonstrate any evidence of bone marrow suppression, as evidenced by the lack of abnormalities in any hematologic parameters during the study that were attributable to abacavir. Additionally, abacavir did not demonstrate any evidence of hepatic, renal, or pancreatic toxicity by biochemical analysis. Most treatment-emergent hematologic and clinical chemistry toxicities were rated as grade 1 or 2. No distinguishing pattern could be detected between the cohorts or treatment arms when clinical chemistry laboratory parameters were examined. The incidence of abnormal laboratory values was generally low, and no dose-related trends were observed. There was no evidence to indicate that abacavir used in combination with zidovudine was associated with an increase in the severity of clinical or laboratory toxicities compared with administration of the study drug alone.57,58
CLINICAL SAFETY AND EFFICACY STUDIES
Studies Conducted in Antiretroviral-Naive HIV-1-Infected Adults
Several phase I/II controlled clinical studies were conducted to determine pharmacokinetics, safety, and antiviral activity of abacavir. A parallel, dose-ranging 12-week study (CNAA2001) was completed in 79 antiretroviral-naive HIV-1-infected adults in the United States with limited prior antiretroviral experience (<12 weeks of zidovudine therapy).57,58 This was the first multiple-dosing study in humans. The doses of abacavir studied were 200 mg tid, 400 mg tid, 600 mg tid, or 300 mg bid. Patients received open-label abacavir monotherapy for 4 weeks, followed by 8 weeks of zidovudine (600 mg/day) or placebo in addition to abacavir. Abacavir given as monotherapy for 4 weeks resulted in median decreases in plasma HIV-1 RNA by 1.11–1.77 log10 copies/mL and median CD4+ T-lymphocyte count increases of 63–111 cells/μL in all groups. At week 12, median plasma HIV-1 RNA levels decreased by 1.02–2.24 log10 copies/mL (for abacavir monotherapy) and by 1.81–2.01 log10 copies/mL (for abacavir plus zidovudine). At week 12, median CD4+ T-lymphocyte counts increased by 79–195 cells/μL (for abacavir monotherapy) and by 93–142 cells/μL (for abacavir plus zidovudine). The percentage of subjects who had plasma HIV-1 RNA levels at week 12 below 400 and 40 copies/mL were 28% and 11%, respectively (abacavir monotherapy) and 69% and 22%, respectively (abacavir plus zidovudine). Eight subjects (10%) discontinued due to adverse events; nausea (n = 4) and hypersensitivity (n = 3) were the most common reasons for withdrawal; there were no fatalities. After 12 weeks of abacavir plus zidovudine or placebo, 72/79 patients were required to interrupt abacavir until essential preclinical studies were completed.
A second randomized, double-blind, dose-ranging clinical trial (CNAB2002) of abacavir 100, 300, and 600 mg bid was conducted in Europe.59 A total of 60 antiretroviral-naive patients with CD4+ T-lymphocyte counts of ≥100 cells/μL and plasma HIV-1 RNA levels of ≥30 000 copies/mL received up to 24 weeks of abacavir therapy alone; 55 then went into an open-label randomized study of abacavir 300 mg bid plus other antiretrovirals for 72 weeks. At week 24, all subjects who remained on abacavir alone were switched to abacavir 300 mg bid/lamivudine 150 mg bid/zidovudine 300 mg bid or other licensed antiretrovirals as determined by their treating physician. At week 4, greater reductions in plasma HIV-1 RNA were seen in subjects receiving 300 or 600 mg abacavir twice daily (median changes −1.55 and −1.61 log10 copies/mL, respectively) than subjects receiving 100 mg abacavir twice daily (median change, −0.63 log10 copies/mL). At 24 weeks, the 300 and 600 mg twice-daily groups had a median change of plasma HIV-1 RNA of −0.70 and −1.30 log10 copies/mL, respectively. During the open-label phase in which zidovudine/lamivudine was added to 300 mg abacavir twice daily, a further median reduction in plasma HIV-1 RNA of 1.74 log10 copies/mL was seen. At week 48, a median 2.82 log10 drop in plasma HIV-1 RNA from randomized baseline was seen in pooled data from all abacavir-treated subjects. Sixty-five percent of patients had a plasma viral load of <400 copies/mL after 48 weeks of antiretroviral therapy (ART) containing abacavir, and 43% of patients had a plasma viral load <50 copies/mL at the same timepoint. An additional decrease in HIV-1 RNA of 2.16 log10 was seen during the open-label phase from the reset open-label combination baseline. Most subjects (46/55, or 87%) received abacavir combined with lamivudine 150 mg bid and zidovudine 300 mg bid since their switch to open label. Overall, this study demonstrated that abacavir 100 mg bid was inferior to 300 mg bid or 600 mg bid, with the latter two doses showing similar viral load reductions. The majority (42/47, 90%) of subjects at week 72 remained on the triple combination of abacavir/lamivudine/zidovudine, four subjects were receiving PIs in addition to the triple combination, and one subject had substituted stavudine for zidovudine. By week 72, the median change in plasma HIV-1 RNA was −2.8 log10 copies/mL. At week 72, 72% of subjects had a plasma HIV-1 RNA <400 copies/mL and 50% of subjects had plasma HIV-1 RNA <50 copies/mL.60 The most frequently seen adverse experiences reported by 55 subjects during the long-term, open-label phase of the study were nausea/vomiting in 23 of the 55 (41%), malaise/fatigue in 10 (18%), headache in nine (16%), muscle pain in six (11%), GI discomfort/pain in seven (12%), sleep disorders in four (7%), and skin rashes in four (7%). These data included new adverse experiences that began once subjects switched to open-label abacavir, and not those that may have been ongoing from the randomized phase.60
The CNAA3003 trial investigated the effect of zidovudine and lamivudine with or without abacavir in antiretroviral-naive adults located in the USA, Spain, Belgium, and the UK.61 This double-blind, randomized phase III study was designed to assess the safety, tolerance, and antiviral activity of abacavir/lamivudine/zidovudine over 16 weeks, as well as the safety, tolerance, and durability of the abacavir/lamivudine/zidovudine response over 48 weeks. Subjects (n = 173 with CD4+ T-lymphocyte counts >100 cells/μL) were randomized 1:1 to receive abacavir 300 mg bid or placebo/lamivudine 150 mg bid/zidovudine 300 mg bid. Subjects were stratified based on plasma HIV-1 RNA at screening: <10 000 copies/mL, 10 000–100 000 copies/mL, or >100 000 copies/mL. Both treatment groups had marked decreases in plasma HIV-1 RNA by week 4, which was sustained in the abacavir/lamivudine/zidovudine group. At week 16, the proportion of subjects with plasma HIV-1 RNA below the limit of detection (<400 copies/mL) was significantly superior in the abacavir/lamivudine/zidovudine group (75%) when compared to the lamivudine/zidovudine group (35%) (P < 0.0001 analyzed by the Cochran–Mantel–Haenzel test controlling for randomized plasma HIV-1 RNA strata). In addition, the triple combination was effective irrespective of the baseline plasma HIV-1 RNA strata. By contrast, the virologic response to lamivudine/zidovudine began to rebound toward baseline until week 16 when subjects were eligible to add abacavir plus other ART. The virologic response to lamivudine/zidovudine at week 16 was also diminished at the higher plasma HIV-1 RNA baseline strata. Mean increase in CD4+ T-lymphocyte counts was similar between the treatment groups at 16 weeks. The triple combination was generally well tolerated (3% of this group withdrew due to adverse experiences as compared to 2.5% of subjects in the lamivudine/zidovudine group).
The CNAF3007/Ecureuil open-label study evaluated the efficacy and safety of the triple nucleoside combination Combivir/abacavir versus Combivir/nelfinavir as first-line therapy in HIV-1-infected adults.62 This randomized, open-label study in 195 HIV-1-infected adults, with plasma HIV-1 RNA 1000–500 000 copies/mL, compared Combivir/abacavir to Combivir/nelfinavir over 48 study weeks. At the intent-to-treat analysis at 48 weeks, 64% and 61% of subjects had plasma viral load <50 copies/mL in the Combivir/abacavir and Combivir/nelfinavir groups, respectively. The baseline viral load was comparable in both groups with a median of 4.2 log10 copies/mL (Combivir/abacavir) and 4.1 log10 copies/mL (Combivir/nelfinavir). The median change from baseline in plasma viral load was −2.3 log10 copies/mL in both groups. Median CD4+ T-lymphocyte count increases were 109 and 120 cells/μL in the Combivir/abacavir and Combivir/nelfinavir arms, respectively. Possible hypersensitivity reactions to abacavir were reported in four subjects (4%). These results suggest that Combivir/abacavir is generally well-tolerated first-line ART in HIV-1-infected adults, with comparable antiviral activity to that of a PI-containing regimen over 48 weeks.
Of 155 subjects completing 48 weeks in the CNAF3007 trial, 92 were enrolled into the CNAF3021 study to look at long-term safety and efficacy: 47 subjects initially randomized to the Combivir/abacavir group and 45 subjects to the Combivir/nelfinavir arm.63 After a follow-up of at least 2 years post-CNAF3007 randomization (median 3 years), there was a higher proportion of patients in the Combivir/nelfinavir group (67%) than in the Combivir/abacavir group (30%) who discontinued the treatment initially attributed by randomization of the CNAF3007. In total, 11/30 patients in the Combivir/nelfinavir switched from nelfinavir to abacavir. In the intent-to-treat analyses (switch included), 76% of subjects in the Combivir/abacavir group and 71% of subjects in the Combivir/nelfinavir group had a plasma HIV-1 RNA <50 copies/mL at the single long-term entry CNAF3021 visit.
The CNA3014 study compared Combivir/abacavir to another PI-containing regimen, indinavir (800 mg tid)/abacavir in a 48-week open-label randomized study in 342 HIV-infected ART naive adults with CD4+ T-lymphocyte counts >100 cells/μL and plasma HIV-1 RNA >5000 copies/mL.64 Subjects were stratified based on screening plasma HIV-1 RNA: >5000–100 000 copies/mL or ≥100 000 copies/mL. Results demonstrated that time to treatment failure over 48 weeks by intent-to-treat analysis was significantly longer in the abacavir/Combivir group than in the indinavir/Combivir group (P < 0.001). In the primary analysis (intent-to-treat, missing = failure, plasma HIV-1 RNA <400 copies/mL), the abacavir/Combivir regimen was superior. In the intent-to-treat analysis at week 48 by baseline plasma HIV-1 RNA stratum, abacavir/Combivir was more effective than indinavir/Combivir in patients with >100 000 copies/mL (70% vs 49% with plasma HIV-1 RNA <400 copies/mL). This difference was not observed in patients with plasma HIV-1 RNA >100 000 copies/mL (60% in abacavir/Combivir vs 51% in indinavir/Combivir). Sustained increases in CD4+ T lymphocytes were observed in both treatment groups over 48 weeks. Self-reported adherence to randomized treatment was significantly higher in the abacavir/Combivir group, with 72% of subjects reporting taking all of their doses or missing an average of less than one dose per week during the last month, compared with 45% of subjects in the indinavir/Combivir group (P < 0.001). The percentages of drug-related adverse events in the study were 65% (108/165 in abacavir/Combivir group) and 87% (142/164 in the indinavir/Combivir group). Six percent (10/165) of the subjects in the abacavir/Combivir group had a possible abacavir hypersensitivity reaction, all of which occurred during the first 6 weeks of therapy.
The KLEAN study examined two other PIs, fosamprenavir–ritonavir versus lopinavir–ritonavir, each in combination with abacavir–lamivudine.65 This open-label, noninferiority study enrolled 878 antiretroviral-naive HIV-1-infected subjects to receive fosamprenavir–ritonavir 700 mg/100 mg twice daily or lopinavir–ritonavir 400 mg/100 mg twice daily, each with the eco-formulation of abacavir–lamivudine 600 gm/300 mg (Epzicom) once daily. Noninferiority of fosamprenavir–ritonavir to lopinavir–ritonavir was demonstrated for the primary study endpoint of proportion of subjects at week 48 achieving plasma HIV-1 RNA <400 copies/mL (95% CI around the treatment difference −4.84–7.05 was shown). At week 48, 315 of 434 (73%) subjects in the fosamprenavir–ritonavir group and 317 of 444 (71%) in the lopinavir-ritonavir group achieved plasma HIV-1 RNA <400 copies/mL. Treatment discontinuations because of an adverse event, which was another primary study endpoint, were few and occurred with similar frequency in the two treatmentgroups (fosamprenavir–ritonavir 53 (12%); lopinavir–ritonavir 43 (10%)). Suspected abacavir hypersensitivity was reported in 53 cases (6%); 32 (7%) in the fosamprenavir–ritonavir arm and 21 (5%) in the lopinavir–ritonavir arm.
Abacavir versus the NNRTI nevirapine combined with zidovudine/lamivudine was examined as a first-line ART for HIV-1 infection in Uganda, Africa.66 The rationale for this safety study was that these agents might avoid drug interactions with tuberculosis therapy as described with HIV-1 PIs, plus the tolerability and toxicity of abacavir in Africa were unknown. This randomized, double-blind, 24-week trial enrolled 599 symptomatic antiretroviral-naive subjects with CD4+ T-lymphocyte counts <200 cells/μL who were either zidovudine/lamivudine (Combivir) with either 300 mg abacavir or nevirapine placebo (n = 300) or nevirapine 200 mg and abacavir placebo (n = 299) twice daily. Of 599 enrolled subjects, 569 (9%) completed 24 weeks; however, 24 (4%) died and six (1%) were lost to follow-up. Twenty subjects had serious adverse reactions (six (2%) on abacavir and 14 (4.7%) on nevirapine) (primary endpoint: hazards ratio = 4.1; 95% CI = 0.16–1.08; log rank P = 0.06). Serious adverse reactions reported in 19 of 20 patients were consistent with potential hypersensitivity reactions: similar proportions on abacavir (four of six) and nevirapine (seven of 13) had respiratory, constitutional (three with abacavir, seven with nevirapine), or GI symptoms (two with abacavir, five with nevirapine), rash (six with abacavir, 10 with nevirapine), fever (six with abacavir, nine with nevirapine), or oral/mucosal involvement (two with abacavir, five with nevirapine); but hepatic involvement was seen only in three patients on nevirapine. The remaining serious adverse reaction was asymptomatic hepatitis (nevirapine arm). In total, 14 (4.7% abacavir) and 30 (10%) nevirapine patients discontinued blinded abacavir/nevirapine (P = 0.01) due to toxicity (six (2%) abacavir vs 15 (5%) nevirapine, respectively, P = 0.05), all rash or possible hypersensitivity reactions (six with abacavir, 13 with nevirapine), or hepatotoxicity (four with nevirapine). This study concluded that abacavir had a lower discontinuation rate and a trend toward a lower rate of serious adverse reactions when compared with nevirapine in African patients initiating ART with low CD4+ T-lymphocyte counts.
The CNA30024 trial studied abacavir versus zidovudine combined with lamivudine and the NNRTI, efavirenz, in an international randomized, double-blind noninferiority clinical trial in 654 antiretroviral-naive HIV-1-infected patients in the USA, Costa Rica, Chile, Portugal, and Denmark.67 Of 654 enrolled subjects, 649 received more than one dose of randomized study medication. A total of 503 (77%) completed ≥48 weeks of study (78% in the abacavir group and 77% in the zidovudine group), and 145 (24%) of the subjects prematurely discontinued the study (similar for both treatment groups). The incidence of reported serious adverse events was 20% in the abacavir group and 14% in the zidovudine group. Of the 30 cases of suspected hypersensitivity reactions reported in the abacavir group, only five met the definition of a serious event. The primary study objective was the comparison of the proportion of subjects achieving plasma HIV-1 RNA ≤50 copies/mL through 48 weeks of study: 70% of subjects in the abacavir arm versus 69% of subjects in the zidovudine group maintained confirmed plasma HIV-1 RNA levels ≤50 copies/mL. Therefore, abacavir was found to be noninferior to zidoduvine when combined with lamivudine and efavirenz (stratified 95% CI of −6.3% to 7.9%). Furthermore, the proportion of subjects with plasma HIV-1 RNA ≤50 copies/mL at week 48 was similar between treatment arms when stratified by screening plasma HIV-1 RNA levels of either <100 000 copies/mL (72% abacavir group vs 70% zidovudine group) or >100 000 copies/mL (67% in both treatment arms).
The ACTG 5095 study compared the triple-nucleoside regimen of abacavir plus Combivir versus NNRTI-containing (efavirenz) regimens for the initial treatment of HIV-1-infection.68,69 This randomized, double-blind study evaluated three antiretroviral regimens for the initial treatment of subjects infected with HIV-1: zidovudine/lamivudine/abacavir, zidovudine/lamivudine plus efavirenz, and zidovudine/lamivudine/abacavir plus efavirenz. Combivir/abacavir (Trizivir) versus Combivir plus efavirenz versus Combivir/abacavir plus efavirenz. A total of 1147 subjects with a mean baseline HIV-1 RNA level of 4.85 log10 copies/mL and a mean CD4+ T-lymphocyte count of 238/μL were enrolled. Prespecified stopping rules led to the recommendation to stop the triple-nucleoside regimen (abacavir plus Combivir) due to virologic inferiority and allowed presentation of results in the triple-nucleoside group in comparison to pooled data from the efavirenz groups.68 The time to virologic failure was found to be significantly shorter in the triple-nucleoside group (P < 0.001). After a median follow-up of 32 weeks, 82 of 382 subjects (21%) in the triple-nucleoside group and 85 of 765 (11%) in the pooled efavirenz groups had virologic failure. These differences persisted despite stratification for baseline plasma HIV-1 RNA levels in subjects either entering the study with <100 000 copies/mL (P = 0.001) or >100 000 copies/mL (P < 0.001). These differences also persisted despite stratification for baseline pretreatment CD4+ T-lymphocyte subjects of ≥100 copies/mL (P = 0.001) or lower (P < 0.001), as well as baseline pretreatment CD4+ T-lymphocyte subjects of ≥200 copies/mL (P = 0.004) or lower (P < 0.001). In a post hoc analysis, the 195 subjects in the triple-nucleoside arm who reported 100% adherence at week 12 had a higher rate of virologic failure than the 382 subjects in the pooled efavirenz arms with the same level of adherence (P < 0.001). Suspected abacavir hypersensitivity reactions were reported in 27 (7%) subjects in the triple-nucleoside group and 59 (8%) subjects in the pooled efavirenz arms. The results of the completed three versus four drug A5095 trial demonstrated no difference between the three-drug regimen of zidovudine/lamuvudine plus efavirenz versus the four-drug regimen of zidvoudine/lamivudine/abacavir plus efavirenz as initial treatment in HIV-1-infected subjects.69 After a median 3 year follow-up, 99 (26%) of 382 and 94 (25%) of 383 patients receiving the three-drug and four-drug regimens, respectively, reached protocol-defined virologic failure; time to virologic failure was not significantly different (hazard ratio, 0.95; 97.5% CI, 0.69–1.33, P = 0.73). Overall, at least 80% of subjects had plasma HIV-1 RNA levels <50 copies/mL through 3 years of treatment. This study demonstrated that adding abacavir as a fourth initial drug to this dual-nucleoside plus NNRTI-containing regimen provided no additional benefit in a direct three versus four initial drugs comparison.
Not all abacavir-containing triple-nucleoside regimens are recommended for HIV-1-infected antiretroviral-naive subjects. Early virologic nonresponse to abacavir, tenofovir, and lamivudine was reported in the ESS30009 Study.70 This randomized open-label, multicenter study compared tenofovir disoproxil fumarate versus the NNRTI efavirenz when both administered once daily with abacavir/lamivudine fixed combination as first-line antiHIV triple-drug therapy. A total of 340 subjects were randomized with a median baseline HIV-1 RNA level of 4.7 log10 copies/mL and median baseline CD4+ T-lymphocyte count of 251 cells/μL. After reports of early virologic nonresponse, an unplanned interim analysis was performed on 194 of the enrolled 340 subjects who had ≥8 weeks of plasma HIV-1 RNA results available. Several definitions of virologic nonresponse were employed: (1) a <2.0 log10 copies/mL decrease in HIV-1 RNA level by study week 8; (2) a rebound of plasma HIV-1 RNA ≥1.0 log10 copies/mL above the nadir; or (3) for subjects with two consecutive plasma HIV-1 RNA measurements <50 copies/mL, a subsequent increase to >400 copies/mL on two consecutive occasions. Interim results revealed that virologic nonresponse occurred in 50 (49%) of 102 subjects in the tenofovir disoproxil fumarate arm when compared to five (5%) of 92 subjects in the efavirenz arm (P < 0.001). By study week 12, genotyping of plasma HIV-1 isolates from nonresponders in the tenofovir disoproxil fumarate/abacavir/lamivudine arm detected the HIV-1 RT M184V or M184I/M/V mixtures in 40 (98%) of 41 subjects and HIV-1 RT K65R and M184V or mixtures in 22 (54%) of 41 subjects (see section on HIV-1 Drug Resistance). Subjects initially randomized to the tenofovir disoproxil fumarate arm were then permitted to switch to an investigator-determined second-line regimen and 131 (77%) of 171 subjects remained in follow-up. Of the 61 subjects with genotype data available at or before switching, 31 had a detectable HIV-1 RT K65R mutation. The efavirenz arm continued unaltered, and by contrast to the abacavir arm, 120 (71%) of 169 subjects achieved a plasma HIV-1 RNA level of <50 copies/mL after 48 study weeks. As mentioned previously (see section on In Vitro Activity and Mechanisms of Action), no drug–drug interactions to explain the virologic nonresponse of abacavir, tenofovir disoproxil fumarate, and lamivudine were found. Rather, the low genetic barrier to development of HIV-1 drug resistance is most likely to blame since this three-drug regimen exerts selective pressure with rapid development of M184V, followed by K65R in some patients. Therefore, the combined regimen of abacavir, tenofovir disoproxil fumarate, and lamivudine for HIV-1-infection should not be used.
The COL40263 trial evaluated the once-daily regimen of trizivir and tenofovir disoproxil fumarate in antiretroviral-naive HIV-1-infected subjects with plasma HIV-1 RNA >30 000 copies/mL.71 The primary endpoints of this open-label, single-arm, multicenter trial were the proportion of subjects with plasma HIV-1 RNA <50 copies/mL at week 48 and the proportion of subjects with grade 3 or 4 adverse events and laboratory toxicities. Of 123 enrolled subjects, 52 (42%) discontinued study through week 48, including 14 due to adverse events, 13 lost to follow-up, and 12 with virologic nonresponse, as well as other reasons. Suspected abacavir hypersensitivity occurred in eight subjects (6.5%) within the first 3 weeks of study drug administration. Of subjects who remained on therapy over 48 weeks, 49 of 64 (77%) of subjects had plasma HIV-1 RNA levels <50 copies/mL at week 48.
The ESS40013 study tested fourdrug induction with abacavir/lamivudine/zidovudine plus the NNRTI efavirenz followed by maintenance treatment with the simplified regimen of abacavir/lamivudine/zidovudine alone in antiretroviral-naive HIV-1-infected subjects.72 All 488 subjects received abacavir/lamivudine/zidovudine plus efavirenz during the 48-week induction phase followed by a 1:1 randomization to either the continued regimen of abacavir/lamivudine/zidovudine plus efavirenz or to simplified regimen of abacavir/lamivudine/zidovudine for the subsequent 48-week maintenance phase. No significant differences were noted in proportion of subjects with plasma HIV-1 RNA level <50 copies/mL at week 96: 79% abacavir/lamivudine/zidovudine plus efavirenz versus 77% abacavir/lamivudine/zidovudine; P = 0.75). Virologic failure occurred in 22 patients during induction and in 24 patients (16 in abacavir/lamivudine/zidovudine arm and eight in abacavir/lamivudine/zidovudine plus efavirenz group; P = 0.134) during maintenance. More subjects reported perfect adherence at week 96 in the triple therapy arm than in the four-drug arm (88.8% vs 79.6%; P = 0.057).

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


