INTRODUCTION
SUMMARY
Diseases of the histiocyte (i.e., macrophage or dendritic cell) lineage can be divided into four groups based upon the final maturation steps from their myeloid progenitor cells: (1) Langerhans cell histiocytosis (LCH), (2) malignant histiocytoses or dendritic cell sarcomas, (3) juvenile xanthogranuloma/Erdheim-Chester disease, Rosai-Dorfman disease, and (4) hemophagocytic lymphohistiocytosis syndromes. Storage diseases of macrophages are discussed in Chap. 72. The distinction among these diseases is based upon clinical characteristics and histopathologic staining for unique surface markers. LCH may present at birth or in adulthood with skin rash, bone pain, draining ears, oral ulcers, gingivitis, pulmonary dysfunction, chronic diarrhea, diabetes insipidus, and marrow or liver failure. Therapy for LCH in children has been studied in clinical trials by the Histiocyte Society. Treatment for adults is based primarily on case series. Although relapses are not typically rapidly fatal, they are associated with a higher risk of endocrine and central nervous system complications. The diagnostic criteria for the malignant histiocytosis have been clarified by cell-surface marker studies. Treatment options and prognosis vary widely. Erdheim-Chester disease and juvenile xanthogranuloma are phenotypically similar, but are treated differently. Erdheim-Chester disease is found almost exclusively in adults and juvenile xanthogranuloma occurs primarily in children. Rosai-Dorfman disease presents with massive cervical lymphadenopathy in most patients, but may also involve other parts of the body. There are several treatment options for Rosai-Dorfman disease, Erdheim-Chester disease, and juvenile xanthogranuloma, but no clinical trials of specific drugs have been published. Hemophagocytic lymphohistiocytosis (HLH) is characterized by pathologic inflammation and may present with infections, hepatitis, meningitis, or autoimmune diseases. Without therapy, HLH is almost universally fatal. Most patients who receive prompt diagnosis and treatment with immune suppression therapy survive.
Acronyms and Abbreviations
AHSCT, allogeneic hematopoietic stem cell transplantation; ALL, acute lymphoblastic leukemia; ATG, antithymocyte globulin; CD, cluster designation; CT, computed tomography; DC, dendritic cell; DI, diabetes insipidus; DLCO, diffusing capacity in lung for carbon dioxide; ECD, Erdheim-Chester disease; FEV1, forced expiratory volume in 1 second; HLA-DR, human leukocyte antigen-D related; HLH, hemophagocytic lymphohistiocytosis; IFN, interferon; IL, interleukin; JXG, juvenile xanthogranuloma; LC, Langerhans cell; LCH, Langerhans cell histiocytosis; M-CSF, macrophage colony-stimulating factor; MRI, magnetic resonance imaging; NK, natural killer; PET, positron emission tomography; RDD, Rosai-Dorfman disease.
CLASSIFICATION OF THE HISTIOCYTOSES
The general description of cells in the monocyte-macrophage system (mononuclear phagocyte system) has been largely clarified (Chaps. 67 to 69), although some ambiguity remains. Nomenclature committees remain loyal to the term “histiocyte,” a designation assigned in the 19th century to tissue macrophages, although the “histiocytosis” umbrella includes functional and neoplastic disorders of a broad range of cells in the monocyte, macrophage, and dendritic cell (DC) lineages. The distinctions among diseases in this category are determined by (1) clinical findings, (2) histopathology, (3) immunocytology to define the antigens on the surface of the pathologic cells, and (4) cytogenetic or genetic features (Table 71–1).
| Histologic Features | LCH | Malignant Histiocytosis | ECD/JXG | HLH | RDD |
|---|---|---|---|---|---|
| HLH-DR | ++ | + | − | + | + |
| CD1a | ++ | +/− | − | − | − |
| CD14 | − | +/− | ++ | ++ | ++ |
| CD68 | +/− | +/− | ++ | ++ | ++ |
| CD163 | − | − | + | ++ | ++ |
| CD207 (Langerin) | +++ | +/− | − | − | − |
| Factor XIIIa | − | − | ++ | − | − |
| Fascin | − | +/− | ++ | +/− | + |
| Birbeck granules | + | +/− | − | − | − |
| Hemophagocytosis | +/− | − | − | +/− | − |
| Emperipolesis | − | − | − | − | + |
The histiocytic disorders have been classified based upon whether they are (1) DC related, (2) monocyte-macrophage related, or (3) malignancies of macrophages or DCs (Table 71–2).1,2 Evolving understanding of myelomonocytic differentiation, as well as cellular origins of histiocytic disorders, will likely necessitate revision of these classifications in the near future.
1. Disorders of varying biologic behavior, lacking cytologic atypia a. Dendritic-cell-related Langerhans cell histiocytosis Juvenile xanthogranuloma Erdheim-Chester disease b. Monocyte-macrophage related Hemophagocytic lymphohistiocytosis Familial and/or with identified dysfunctional gene mutation Secondary hemophagocytic syndromes Infection-associated Malignancy-associated Autoimmune-associated Other Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) Solitary histiocytoma of macrophage phenotype 2. Malignant disorders Dendritic cell related Histiocytic sarcoma Monocyte-macrophage related Leukemias: monocytic M5A and M5B, myelomonocytic M4, chronic myelomonocytic leukemia |
The pathologic cells in Langerhans cell histiocytosis (LCH) lesions have phenotypic similarity to epidermal Langerhans cells (LCs), which has led to the hypothesis that LCH is derived from aberrant activation and/or neoplastic transformation of the epidermal LCs.3 LCH originates from aberrant proliferation and differentiation of myelomonocytic precursors.4,5 Regardless of ontogeny, LCH lesions are characterized by pathologic DCs with phenotypic similarity to epidermal LCs, including positive staining with anti-CD207 (antilangerin) and the presence of Birbeck granules identified by electron microscopy.6,7 Birbeck granules are racket-shaped inclusions that are thought to be involved in antigen processing. Cells staining with anti-CD207 and/or anti-CD1a are required for the diagnosis of LCH. Other antigens, such as S100 or HLA-DR (human leukocyte antigen-D related) are not specific for LCH. The histiocytes in Erdheim-Chester disease (ECD) and juvenile xanthogranuloma (JXG) have phenotypic similarity to the dermal–interstitial dendrocyte that stains with antibodies to CD68, fascin, and factor XIIIa. However, the DCs in these disorders also express surface CD163 that is characteristic of macrophages.
Malignant histiocytosis (or “histiocytic sarcoma”) has evolved as a diagnosis of exclusion involving malignant histiocytes that lack markers for anaplastic large cell lymphoma or other hematologic malignancies. Malignant histiocytosis represents a spectrum of malignancies that presumably derive from DCs of different lineages at different stages of differentiation. Human DCs are defined as hematopoietic cells expressing high levels of major histocompatibility complex II (MCHII) and CD11c while lacking other specific lineage markers. Under normal conditions, these cells reside in tissue or circulate in the blood. Once stimulated by antigen, they migrate to lymphoid tissue and interact with effector or suppressor T cells. Malignant histiocytoses are variable and share surface markers with DC subsets: follicular DC “sarcoma” (CD21+, CD35+), interdigitating DC “sarcoma” (CD14+), and Langerhans cell “sarcoma” (CD1a+).
The monocyte-macrophage disorders include Rosai-Dorfman disease (RDD) and hemophagocytic lymphohistiocytosis (HLH). RDD, also known as sinus histiocytosis with massive lymphadenopathy, has the telltale histopathologic finding of intact lymphocytes in the cytoplasm of macrophages (emperipolesis), a feature that must be present to diagnose this disorder. HLH is distinct among the diseases discussed in this chapter in that the macrophages are nonneoplastic, otherwise normal histiocytes, characterized by a pathologic reaction to aberrant stimuli.
LANGERHANS CELL HISTIOCYTOSIS
LCH has a complicated history that underlies the current clinicopathologic approach to the disease. What has come to be identified as LCH was first described in case reports and series in the early 1900s.8 By the 1950s, patterns of clinical presentations had been categorized as Hand-Schüller-Christian (multifocal eosinophilic granulomas) and Letterer-Siwe (disseminated disease including marrow, spleen and liver). However, these apparently disparate entities were found to share the same histopathology: histiocytes with abundant cytoplasm and reniform nuclei among an inflammatory infiltrate that could include lymphocytes, eosinophils and macrophages. Lichtenstein hypothesized that these clinical disorders must be linked by a common etiology, and proposed the designation “Histiocytosis X,” with “X” indicating incomplete knowledge of pathogenesis and cell of origin. Two decades later, Birbeck granules, which had previously been identified only in epidermal LCs, were identified in DCs of LCH lesions by electron microscopy. Nezelof and colleagues therefore extended Lichtenstein’s hypothesis that this spectrum of disorders arises from the epidermal Langerhans cell.3 Histiocytosis X has since been regarded as “Langerhans cell histiocytosis.”
The incidence of LCH is 2 to 10 cases per 1 million children younger than age 15 years.9,10,11 A survey of LCH patients in France revealed an incidence of 4 to 6 per 1 million in children younger than age 15 years. The male-to-female ratio is close to 1 and the median age of presentation is 30 months, although patients may present with the disease from birth through the ninth decade. Identical and fraternal twins with early onset of LCH have been described. There are occasional reports of affected nontwin siblings and multiple cases in one family, although it is not clear if this is significantly greater than one would expect by chance.12 The relatively high rate of high-risk multisystem LCH in identical twins compared to presumed fraternal twins may be explained by shared precursor cells as well as the possibility of shared genes. An increased frequency of family members with thyroid disease,13 family members with other cancers,14 in vitro fertilization,15 and parental exposure to metal16 have also been reported as potential associations. Although inheritance of penetrant mendelian “LCH genes” in the majority of cases seems unlikely, it remains possible that there are inherited genes associated with increased risk of developing LCH.
The focus of studies and reviews on LCH over the past decades has been on either an immune or a neoplastic disorder. The competing models have been (1) an inappropriate activation of an otherwise normal epidermal LC or (2) a neoplastic transformation of the epidermal LC. Twenty years ago, CD1a+ cells from LCH lesions were described as clonal, based on non–random X inactivation.17,18 Subsequently, somatic activating mutations in the BRAF oncogene were reported in 57 percent of LCH histopathologic specimens,19 with subsequent studies validating the recurrent BRAFV600E mutation at high frequency.5,20,21,22 BRAF is the central kinase of the RAS/RAF/MEK/ERK pathway, which is essential to numerous cell functions and is frequently mutated in cancer cells.23 Significance of BRAFV600E as a driver mutation in LCH is supported by early reports of clinical responses to BRAF inhibition in adults with combined LCH and ECD.24 Other recurrent somatic mutations in LCH may be uncovered.
The cell of origin of LCH has been assumed to be the epidermal LC based on phenotypic similarities discussed above. However, the transcriptome of CD207+ cells from LCH lesions is more consistent with an immature myeloid DC phenotype than with the transcriptome of epidermal LCs.4 Furthermore, DC maturation may be heterogeneous within lesions, with variable CD1a+/CD207− populations.25,26 Immuno-histochemical staining antibodies specific for BRAFV600E revealed that the mutations are not limited to CD207+ cells within LCH lesions, but also are found in CD207-negative subpopulations.21 Using the BRAFV600E mutation as a “bar code,” cells that carry the mutation were identified in circulating myelomonocytic precursors in blood and in hematopoietic stem cells in marrow aspirates of patients with clinical high-risk LCH, but not in patients with single-lesion low-risk LCH. The functional significance of this observation was supported by the ability of forced expression of BRAFV600E in myelomonocytic precursors (CD11c+ cells) to induce a disseminated LCH-like phenotype in mice.5 We therefore hypothesize that the state of differentiation of the cell in which LCH arises determines the clinical manifestations of the disease; pathologic ERK (extracellular signal-regulated kinase) activation in stem cell or early myelomonocytic precursor resulted in disseminated high-risk disease whereas ERK activation in tissue-restricted precursor resulted in localized disease. These observations define LCH as a myeloid neoplasm.
Although ERK hyperactivation may drive differentiation and proliferation of myelomonocytic precursors in LCH, the mechanisms that drive inflammation in the LCH lesions are not currently understood. The LCH DCs make up a median of 8 percent of the cells within lesions.5 Like physiologically activated DCs, they express high levels of T-cell costimulatory molecules and proinflammatory cytokines.4,27,28 The LCH lesion’s inflammatory infiltrate includes lymphocytes, macrophages, and eosinophils in variable proportions, with enrichment of regulatory CD4+CD25+ T cells (T regs).29 Dozens of cytokines, chemokines, and cytokine and chemokine receptors have been hypothesized to play roles in LCH pathogenesis, creating a local “cytokine storm” as well as increased circulating proinflammatory cytokines, including tumor necrosis factor α, soluble interleukin (IL)-2 receptor α, RANKL (receptor activator of nuclear factor-κB ligand), osteoprotegerin, and osteopontin.4,30,31 Although MAPK (mitogen-activated protein kinase) pathway activation in maturing DCs may drive differentiation of LCH DCs, inflammation likely plays a role in the clinical manifestations and possibly also in tumor maintenance: A unique phenomenon in LCH is that disruption of solitary LCH lesions often results in spontaneous resolution, even without clean margins.
LCH usually presents with a skin rash or painful bone lesion. Systemic symptoms of fever, weight loss, diarrhea, edema, dyspnea, polydipsia, and polyuria may also occur.
In LCH, involvement of specific organs at the time of diagnosis determines the designation “high-risk” or “low-risk.” Organs that indicate high-risk of progression include liver, spleen, and marrow. Organs that indicate low-risk of progression include skin, bone, lung, lymph nodes, and pituitary gland. Patients may present with disease in one site or organ (single site or single system) or in multiple sites or organs (multisystem). Treatment decisions for patients are based on whether or not organs that indicate high-risk or low-risk of progression are involved, and if LCH presents as a single site or as a multisystem disease. Patients can have LCH of the skin, bone, lymph nodes, and pituitary in any combination and still be considered to have a low-risk of progression.
In this situation the disease presents with involvement of one site, which can be skin, oral mucosa, bone, lymph nodes, pituitary, or thymus.
Skin Lesions simulating seborrheic dermatitis of the scalp may be mistaken for prolonged “cradle cap” in infants. The lesions may be localized to intertriginous areas or may be diffuse (Fig. 71–1). The most common skin flexures affected are the groin, the perianal area, back of the ears, the neck, the armpits, and, in women, the crease below the breasts. Infants may also present with brown to purplish papules over any part of their body. This latter manifestation may be self-limited as the lesions often disappear during the first year of life with no therapy. However, these patients should be evaluated for other sites of disease which may coexist with skin lesions as multisystem LCH. Some reports describe multisystem LCH arising after presenting with lesions limited to the skin,32,33 although infants with skin lesions were not observed to develop disseminated disease in a relatively large institutional series.34 In a report of 61 neonatal LCH cases, nearly 60 percent had multisystem disease and 72 percent had high-risk organ involvement.35 The overall survival was poorer in neonates with high-risk organ involvement compared to infants and children with the same extent of disease. Response to therapy at 12 weeks was more important than patient age in determining outcome. A review of 71 children with skin involvement revealed that those without other organ systems involved had nearly a 90 percent progression-free survival after initial therapy or observation. Often dermatologists or general practitioners who see a patient with skin LCH do not perform a complete evaluation searching for LCH in other sites. Thus, we found that 40% of patients referred for apparent “skin only” LCH did have other sites of disease when a complete evaluation was done.34
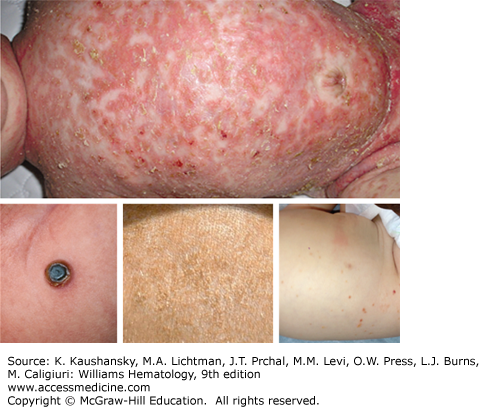
Isolated skin involvement is rarely observed in children older than 18 months of age.34 Children and adults may develop red papular lesions in the scalp, skin of the groin, abdomen, back, or chest that resemble the diffuse rash of Candida infection. Seborrhea-like involvement of the scalp may be mistaken for a severe case of dandruff in older individuals. Ulcerative lesions behind the ears, involving the scalp, skin of the genitalia, or perianal region are often misdiagnosed as bacterial or fungal infections.
Oral Mucosa Presenting symptoms include gingival hypertrophy, ulcers of the soft or hard palate, buccal mucosa, or on the tongue and lips. Lesions of the oral mucosa may precede evidence of LCH elsewhere.36
Bone The most frequent site of LCH in children is a lytic lesion of the skull which may be asymptomatic or painful.37 LCH can occur in any bone. The most frequently involved sites are skull, femur, ribs, vertebrae, and humerus. Spine lesions are most often located in the cervical vertebrae and are frequently associated with other bone lesions. Proptosis from a LCH mass in the orbit mimics rhabdomyosarcoma, neuroblastoma, and benign fatty tumors of the eye. Some skull lesions are not only lytic but may have an accompanying mass that impinges on the dura. Whether or not this affects risk of progression or not is unknown. Lesions of the facial bones (orbit, mastoid), or anterior or middle cranial fossae (e.g., temporal, sphenoid, ethmoid, or zygomatic bone) comprise the “CNS-risk” sites. These patients have a threefold increased risk for developing diabetes insipidus (DI) and an increased risk of other CNS disease (see “Central Nervous System and Endocrine System” below).
Lymph Nodes and Thymus Cervical nodes are the ones most frequently involved and may be soft or hard-matted masses with accompanying lymphedema. An enlarged thymus or mediastinal node involvement can mimic lymphoma or an infectious process and may cause asthma-like symptoms. Biopsy of the node or mass with histologic examination and microbial cultures is helpful even in patients with known LCH as lymphadenopathy may represent LCH, coexisting neoplastic disease, or infection.38
Pituitary Gland The posterior pituitary gland can be affected in LCH patients causing central DI (see “Endocrine System” below). Anterior pituitary involvement may result in impaired growth and sexual maturation.
In multisystem LCH, the disease presents in multiple organs or body systems, including liver and spleen, marrow (high-risk sites) or bones, lungs, skin, lymph nodes endocrine system, gastrointestinal system (low-risk sites), and CNS (intermediate-risk site depending on extent).
Liver and Spleen Patients with liver and spleen involvement have a significantly increased risk of death from LCH. Hence, these are considered “high-risk organs.”39 Hepatic infiltration by LCH lesions can be accompanied by dysfunction, leading to hypoalbuminemia with ascites, hyperbilirubinemia, and clotting factor deficiencies. Sonographic imaging, computed tomography (CT), or magnetic resonance imaging (MRI) of the liver may show hypoechoic or low-signal intensity along the portal veins or biliary tracts when the liver is involved.40 One of the most serious complications of hepatic LCH is cholestasis and progressive sclerosing cholangitis.41 The median age of children with hepatic LCH is 23 months. Patients generally present with hepatomegaly with or without splenomegaly, elevated alkaline phosphatase, liver transaminases, and γ-glutamyl transpeptidase. Biopsies may or may not show CD207+ DCs.42 A classic histologic feature is the collection of lymphocytes around bile ducts. Seventy-five percent of children with sclerosing cholangitis do not respond to chemotherapy and ultimately require liver transplantation.41
Massive splenomegaly may lead to consumptive cytopenias and respiratory compromise. Splenectomy may ameliorate severe thrombocytopenia, but the effect is generally not sustained with progressive hepatomegaly and inflammation in the setting of uncontrolled disseminated LCH.
Lung The lungs were once considered a high-risk organ; however, review of a large series of patients shows that the treatment outcome for patients with lung and bone involvement is not statistically different from those with only bone LCH.43 The lungs are less frequently involved in children (13 percent) than in adults (60 percent), in whom smoking is a key etiologic factor.44 In young children with diffuse disease, therapy can halt tissue destruction and normal repair mechanisms may restore some lung parenchyma and function. “Spontaneous” pneumothorax can be the first sign of LCH in the lung. Patients also present with cough, tachypnea, or dyspnea. Ultimately, widespread fibrosis and destruction of lung tissue leads to severe pulmonary insufficiency. Declining diffusion capacity may also herald the onset of pulmonary hypertension.45 Chest radiographs may show a nonspecific interstitial infiltrate. A high-resolution CT image of the chest is needed to visualize the cystic and nodular pattern of LCH that leads to the destruction of lung tissue.
Marrow Involvement of the marrow is considered an indicator of high risk. Most patients with marrow involvement are young children who also have diffuse disease in the liver, spleen, lymph nodes, and skin with significant thrombocytopenia or neutropenia, though some may also have scattered marrow involvement with more mild cytopenias.46,47 An institutional study found patients with high-risk LCH with the BRAFV600E mutation to have 0.2 to 2.1 percent of cells from the marrow aspirate carry the mutation, with only four of seven cases being reported as having abnormal histology.5 LCH patients sometimes present with hemophagocytosis in the marrow.48 The presence of CD1a+/CD207+ in the marrow or at other sites identifies hemophagocytic syndrome as secondary to LCH rather than from primary HLH (discussed in the section “Hemophagocytic Lymphohistiocytosis” below).
Endocrine System DI is the most frequent endocrine manifestation of LCH. Patients may present with an apparent “idiopathic” DI and sometimes with an enlarged pituitary gland or stalk before other LCH lesions are identified. Approximately half of these patients will have other lesions diagnostic of LCH within a year of identifying the DI.49 A review of patients with DI and enlarged pituitary glands found the three most likely diagnoses were germinoma, LCH, and lymphoma.50 DI followed the initial LCH diagnosis at other sites by a mean of 1 year and growth hormone deficiency occurred on the average 5 years later. Historically, the 10-year risk of pituitary involvement has been reported as 24 percent.51 In one series, this incidence of DI did not decrease in chemotherapy-treated patients (see “Central Nervous System and Diabetes Insipidus” below). In another study the incidence of DI decreased from 40 to 20 percent after 6 months of treatment with vinblastine and prednisone for patients at risk for CNS involvement.52 However, after a year of this treatment, the incidence of DI was decreased to 12 percent.39
Craniofacial Lesions Patients with multisystem disease and craniofacial involvement at the time of diagnosis, particularly of the ear, eye, and oral region, carried a significantly increased risk of developing DI (relative risk: 4.6).53 This risk increased when the disease remained active for a longer period of time or reactivated. The risk for development of DI in this population was 20 percent at 15 years after diagnosis. Up to 56 percent of DI patients will develop anterior pituitary hormone deficiencies (growth, thyroid, or gonad-stimulating hormones) within 10 years of the onset of DI.54
Gastrointestinal System A few patients with diarrhea, hematochezia, perianal fistulas, or malabsorption have been reported.55,56 Diagnosing gastrointestinal lesions in LCH is difficult because of the patchy involvement. Endoscopic evaluation may reveal CD1a+/CD207+ cells in the intestinal mucosa, though LCH involvement may be patchy and require multiple biopsies to detect.
Central Nervous System and Diabetes Insipidus DI (considered both an endocrine and a CNS manifestation of LCH) can present as an early or late condition. DI caused by damage to the posterior pituitary is the most frequent initial sign (and early manifestation) of LCH in the CNS. Pituitary biopsies are rarely done and only if the stalk is larger than 6.5 mm or there is a hypothalamic mass. The pituitary enlargement may spontaneously decrease or respond to chemotherapy.57 However, a review of 22 patients with pituitary enlargement of 6.5 mm or greater revealed that despite regression of the mass with therapy, all had anterior pituitary deficiencies as well as MRI evidence of the CNS neurodegenerative syndrome (see “Other Chronic Central Nervous System Disease Manifestations” below) and 17 (77 percent) developed clinical signs of neurodegeneration.58 Most often the diagnosis of LCH is established by biopsy of skin, bone, or lymph node of a patient who also has the pituitary abnormalities.
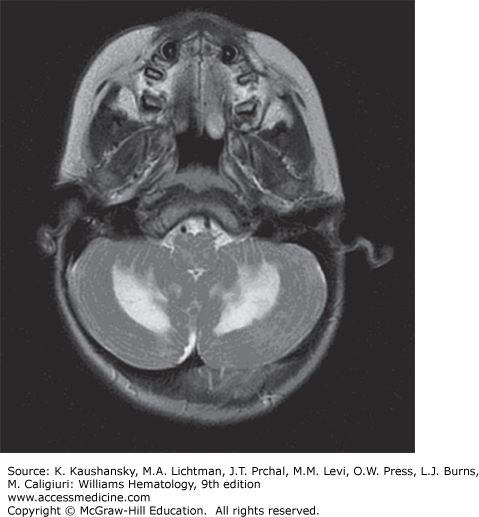
Other Chronic Central Nervous System Disease Manifestations LCH patients may develop mass lesions of the choroid plexus, or gray or white matter.59 These lesions may contain CD1a+ DCs as well as CD8+ lymphocytes.60 A chronic CNS problem that develops in 1 to 4 percent of LCH patients is the “LCH CNS neurodegenerative syndrome” manifested by dysarthria, ataxia, dysmetria, and, sometimes, behavior changes.61,62 The brain MRI in these patients shows hyperintensity of the dentate nucleus and white matter of the cerebellum on fluid-attenuated inversion recovery (FLAIR) and T2-weighted images or hyperintense lesions of the basal ganglia on T1-weighted images (Fig. 71–2). Atrophy of the cerebellum also may be seen.59 The radiologic findings may precede the onset of symptoms by many years or can be found coincidently. Among 83 LCH patients who had at least two MRI studies of the brain for evaluation of craniofacial lesions, DI, other endocrine deficiencies, or neuropsychological symptoms, 57 percent had radiologic neurodegenerative changes at a median time of 34 months after diagnosis. Of these patients, one-quarter had clinical neurologic deficits develop 3 to 15 years after LCH diagnosis.62
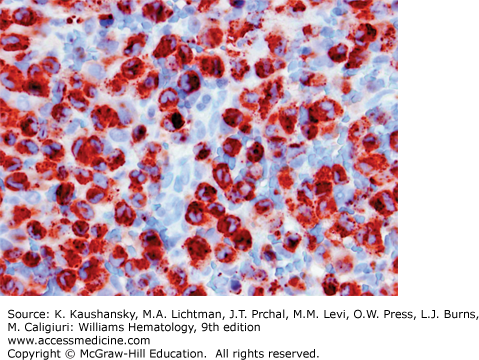
LCH is defined by characteristic inflammatory lesions including histiocytes expressing CD1a and CD207 (Fig. 71–3).1 Patients with high-risk disease may present with anemia and thrombocytopenia caused by marrow involvement and/or inflammation.46,47 An elevated sedimentation rate and thrombocytosis has been reported to correlate with active LCH, but these associations are variable.63 When the liver is involved hypoalbuminemia, elevated liver enzymes, and elevated bilirubin may be observed. Intestinal involvement may also cause hypoalbuminemia. Lytic lesions of the bone are identified by plain films, CT imaging, MRI, bone scan, or positron emission tomography (PET) scan. PET scans are useful for detecting lesions not found by bone scan or plain films and comparison PET scans are particularly good for providing evidence of healing after 6 to 12 weeks of therapy.64 An institutional series found the presence of the somatic mutation in LCH lesions to correlate with higher risk of initial treatment failure, and circulating cells with the BRAFV600E mutation to correlate with active disease, though these observations remain to be validated in prospective trials.5
It is not unusual for infants with LCH skin lesions to have symptoms for longer than 1 year prior to having a diagnostic biopsy.34 The varied cutaneous presentations of LCH may mimic a fungal diaper rash, seborrheic scalp rash or cradle cap, congenital viral infections, neuroblastoma, contact dermatitis, or psoriasis (see Fig. 71–1). Women or men with genital lesions may be thought to have a sexually transmitted disease or other infection. Oral lesions mimic other ulcerative conditions, gingival infections, or dental caries. Copious white or green discharge from the ears resembles otitis externa. Lytic bone lesions are often thought to be evidence of a malignancy such as neuroblastoma, rhabdomyosarcoma, or Ewing sarcoma. Collapsed vertebrae from LCH may mimic tuberculosis bone disease, trauma, or osteomyelitis. The interstitial infiltrates found in LCH patients with pulmonary involvement may resemble a viral pneumonia. An enlarged thymus or mediastinal lymph nodes can cause respiratory distress and wheezing similar to asthma. Enlarged lymph nodes from LCH mimic any infiltrative condition such as lymphomas, other histiocytic diseases, infections, or immune-related conditions. Likewise, hepatosplenomegaly of LCH patients can result from the same conditions. Chronic diarrhea in LCH patients may initially be considered to be an infectious or inflammatory bowel disease. Isolated DI with enlargement of the pituitary may suggest a germinoma, lymphoma, or hypophysitis. LCH should be strongly considered when symptoms of other more common conditions do not respond to therapy.
Occasionally infiltrates of LC are found in various malignancies and as such represent an attempt of the immune system to respond to that disease.65,66,67 Similarly LCH may be found in the thymus of patients with myasthenia gravis.68
The current optimal treatment of childhood and adults LCH patients, as with other rare conditions, is on clinical trials. The U.S. National Cancer Institute website (http://www.cancer.gov/cancertopics/pdq/treatment/lchistio/HealthProfessional) and the Histiocytosis Association (http://www.histio.org; 1–856–589–6606) also may be useful resources.
Patients with only skin LCH, some single-bone lesions (non–CNS-risk), and isolated DI have not been studied in Histiocyte Society clinical trials, but are discussed in this section.
Skin-Limited Lesions Skin-limited lesions may require therapy if they are symptomatic. One approach is topical glucocorticoids,33 although rarely effective. Other approaches with reported efficacy include oral methotrexate (20 mg/m2 weekly)69 or oral thalidomide (50 to 200 mg daily).70 Topical application of nitrogen mustard may be effective for cutaneous LCH that is resistant to oral therapies, but is contraindicated for large areas of skin.71 Psoralen plus long-wave ultraviolet A radiation (PUVA) has been used as well.72 The approach is limited by the severity and distribution of the skin involvement.
Single Skull Lesions of the Frontal, Parietal, or Occipital Regions, or Single Lesions of Any Other Bone Curettage or curettage plus injection of methylprednisolone may be used.73
Vertebral or Femoral Bone Lesions at Risk for Collapse Isolated radiation therapy is indicated for patients with single bone lesions of a vertebrae or the femoral neck, which are at risk of collapse.74,75 When instability of the cervical vertebrae and neurologic symptoms are present, bracing or spinal fusion may be needed.76 Certain skull lesions, not in the CNS-risk region, could also be considered for radiation therapy.
Skull Lesions in the Mastoid, Temporal, Orbital or Base of Skull Bones (CNS-Risk Lesions) The purpose of treating these patients with systemic therapy is to decrease the risk of developing DI. In a large series of patients with CNS-risk lesions who received little or no chemotherapy there was a 20 to 50 percent incidence of DI compared to incidence rates of 10 percent in patients treated with systemic chemotherapy.52 The current standard of care, based on the LCH-III study, is to treat patients with single or multifocal lesions in CNS risk sites for 12 months with intravenous vinblastine and oral prednisone: weekly intravenous vinblastine (6 mg/m2) for 7 weeks, with daily oral prednisone (40 mg/m2) for 4 weeks followed by a 2-week taper. If there is a good response by 6 weeks, then vinblastine frequency is decreased to every 3 weeks. After the first 6 weeks, oral prednisone is given for 5 days at 40 mg/m2 every 3 weeks with the vinblastine intravenous. Patients with suboptimal responses by 6 weeks are given an additional 6 weeks of weekly intravenous vinblastine.39
Multiple Bone Lesions or Combinations of Skin, Lymph Node, or Pituitary Gland Involvement with or Without Bone Lesions Patients should be treated for 12 months with intravenous vinblastine and oral prednisone as outlined for the CNS-risk lesions. Both shorter (<6 months) treatment strategies and treatment with only a single agent (e.g., prednisone) are associated with inferior outcomes. A 37 percent reactivation rate with a two-drug regimen has been reported, versus 50 to 80 percent with only surgery or single-drug treatments.39,53,77
Spleen, Liver, or Marrow (May or May Not Include Skin, Bone, Lymph Node, or Pituitary Gland Involvement) The standard therapy used for LCH in high-risk organs (spleen, liver, and/or marrow) is based upon the LCH-III results in which oral mercaptopurine was added to intravenous vinblastine/oral prednisone regimen, outlined for the low-risk patients above.39 The addition of oral or intravenous methotrexate did not improve the overall survival or risk of reactivation. Another more intensive regimen (JLSG-96) has been reported that includes intravenous cytarabine, intravenous vincristine, oral prednisolone, and methotrexate for good responders or a salvage therapy with intravenous daunorubicin, intravenous cyclophosphamide, intravenous vincristine, and oral prednisolone for poor responders.78 Both treatments lasted 7.5 months. Table 71–3 compares the results of the LCH-III and JLSG-96 trials.
Central Nervous System Treatment of mass lesions, including enlargement of the hypothalamic–pituitary axis, parenchymal mass lesions, and leptomeningeal involvement, with cladribine has been effective. Doses of cladribine ranged from 5 to 13 mg/m2 given intravenously at varying frequencies.79
Reports for treatment of the CNS neurodegenerative syndrome have been limited to case series and pilot studies. Strategies include oral dexamethasone, intravenous cladribine, oral all-trans retinoic acid, intravenous immunoglobulin, and intravenous cytarabine.80,81,82 All-trans retinoic acid was given at a dose of 45 mg/m2 daily, orally, for 6 weeks, then 2 weeks a month for 1 year. Intravenous immunoglobulin, 400 mg/m2 monthly. Some investigators have given chemotherapy along with the IVIG. The chemotherapy plan was prednisolone 2 mg/kg 5 days a month with or without oral or IV methotrexate 20 mg/m2 one day every 2 weeks, daily oral 6-mercaptopurine 1.5 mg/kg/day, or monthly vinblastine 6 mg/m2/dose once monthly for a year. MRI findings were stable, but clinical efficacy was difficult to judge as patients were reported to have no progression in their neurologic symptoms. Intravenous cytarabine with or without intravenous vincristine was effective in decreasing neurologic symptoms and improving the MRI images in five of eight patients who were stable for longer than 8 years.
Recurrent “Low-Risk” Organ Involvement The optimal therapy for patients with relapsed or recurrent disease has not been determined. Patients with recurrent bone disease who reoccur more than 6 months after stopping vinblastine and prednisone can benefit from treatment with a “reinduction” of intravenous vinblastine, weekly, and daily oral prednisone for 6 weeks. If there is no active disease, or at least very little evidence of active disease, then treatment can be changed to every 3 weeks with the addition of oral methotrexate weekly and oral 6-mercaptopurine nightly. Three approaches (1) intravenous cladribine, (2) intravenous vincristine and intravenous cytarabine, and (3) intravenous clofarabine are effective regimens for patients with recurrent bone disease.83,84,85
A phase II trial of oral thalidomide for LCH patients (10 low-risk patients; 6 high-risk patients) who failed primary and at least one secondary regimen showed a complete response in 4 of 10 and partial responses in 3 of 10 low-risk patients. However, dose-limiting toxicities and extraordinary cost limit the overall usefulness of thalidomide.70
Recurrent High-Risk Organ Involvement A new treatment plan is indicated when a patient with multisystem involvement progresses after 6 weeks of standard treatment, or has not had a partial response by 12 weeks. Data from the LCH-III study showed only 57 percent survival for this group. A prospective trial with cladribine (5 mg/m2 per day, intravenously, for 5 days monthly for 6 months) had high rates of response in patients with recurrent and refractory low-risk, but not high-risk, LCH.85 Another regimen for patients with refractory high-risk LCH is treatment with a highly intensive strategy based on acute myelogenous leukemia protocols. Treatment with high-dose cladribine (9 mg/m2 per day) coupled with cytarabine (1 g/m2 per day) for 5 days for at least 2 months resulted in increased overall survival and cure in previously refractory patients. However, there was also a relatively high treatment-related mortality.86 Institutional case series with intravenous clofarabine (25 mg/m2/day for 5 days/month) have promising results in patients who have failed multiple previous strategies with manageable toxicities.87,88,89 There is considerable interest in use of the BRAF inhibitors for treatment of refractory and recurrent LCH in patients with the BRAFV600E mutation. There are pediatric and phase I studies for several anti-BRAF compounds. Concern for more widespread use in the setting of pediatric LCH is tempered by of the toxicity profile that includes high incidence of squamous cell carcinoma in melanoma patients.90 There is only one published report on use of oral vemurafenib for two adult patients who had LCH and ECD with positive responses.91
Allogeneic hematopoietic stem cell transplantation (AHSCT) has been used for patients with multisystem high-risk organ involvement that is refractory to chemotherapy. Reduced-intensity conditioning regimens are curative and associated with less toxicity and improved survival.92
Options No Longer Considered Effective Treatments for LCH in any location that have been used in the past but are no longer recommended include cyclosporine and interferon (IFN)-α. Extensive surgery is also not indicated. It is critical that LCH bone lesions (especially skull and mandible) not be treated by excision with wide margins. Even large defects may remodel to near normal architecture following resolution of LCH follow chemotherapy and/or curettage. Similarly, surgical resection or radiotherapy of groin or genital lesions is contraindicated as chemotherapy can heal skin lesions.
LCH patients with low-risk disease treated with vinblastine and prednisone have a 99 percent chance of survival, but more than 50 percent fail to be cured with initial therapy. Nearly 100 percent of these patients are ultimately cured of LCH, though many require multiple courses of salvage therapy. Patients with high-risk disease who do not respond adequately by 12 weeks of treatment now have an 87 percent chance of long-term survival, but also often require salvage therapy as described earlier.
Disease-associated sequelae remain a major challenge for patients with LCH, with risk of complications increasing with multisystem, high-risk, and prolonged duration of active disease. Children with low-risk organ involvement (skin, bones, lymph nodes, or pituitary gland) treated for 6 months have a 24 percent chance of developing long-term sequelae.93 Those with DI are at risk for panhypopituitarism and should be monitored carefully for adequacy of growth and development. In a retrospective review of 141 patients with LCH and DI, 43 percent developed growth hormone deficiency.54 The 5- and 10-year risks of growth hormone deficiency among children with LCH and DI were 35 percent and 54 percent, respectively. There was no increased reactivation of LCH in patients who received replacement growth hormone compared to those who did not.
Patients treated before 2000 with multisystem involvement had a 71 percent incidence of long-term problems.93,94 Hearing loss has been found in 13 percent of children treated for LCH. Neurologic symptoms secondary to vertebral compression of cervical lesions have been reported in LCH patients with spinal lesions. Cognitive defects and MRI abnormalities may develop in some long-term survivors with CNS-risk skull lesions.95 Some patients have markedly abnormal cerebellar function and behavior abnormalities, while others have subtle deficits in brain stem–evoked potentials and short-term memory.96
Orthopedic problems from lesions of the spine, femur, tibia, or humerus may be seen in 20 percent of patients. These problems include vertebral collapse or instability of the spine that may lead to scoliosis, and facial or limb asymmetry.
Diffuse pulmonary disease may result in poor lung function with higher risk for infections and decreased exercise tolerance. These patients should be followed with pulmonary function testing including the diffusing capacity of carbon monoxide and ratio of residual volume to total lung capacity.45
Liver disease may lead to sclerosing cholangitis which responds to chemotherapy in only 25 percent of cases and liver transplantation usually is indicated.41
Dental problems characterized by loss of teeth have been significant for some patients, usually related to overly aggressive dental surgery.93
Marrow failure secondary to LCH or from therapy is rare but is associated with a higher risk of malignancy. Patients with LCH may have a higher than normal risk of developing secondary cancers.97 Leukemia (usually acute myelogenous) occurs after treatment as does lymphoblastic lymphoma. Concurrent LCH and a malignancy have been reported in a few patients, and some patients have had their malignancy initially followed by development of LCH. Three patients with T-cell acute lymphoblastic leukemia (T-ALL) and aggressive LCH, for which the two disorders had shared clonal markers have been reported.98,99 Two cases had clonality of the same T-cell receptor genotype. The authors considered the plasticity of lymphocytes permitting development into LCs. The other patient with LCH after T-ALL had the same T-cell receptor gene rearrangements and activating mutations of the NOTCH1 gene in the patient’s DCs and acute lymphoblastic leukemia (ALL) blasts. A series of four patients with acute leukemia of ambiguous or myeloid lineage with intermingling LCH cells have been reported.100 The authors speculated the two diseases shared a common hematopoietic stem cell as had been suggested earlier during investigation of the BRAFV600E mutation in the marrow of LCH patients.6
It is estimated that one to two adult cases of LCH occur per 1 million population.101 The true incidence of this disease is difficult to assess because large published studies usually are from referral centers and the disorder often is underdiagnosed. A survey from Germany reported that 66 percent of the adult LCH patients were women with an average age of 43.5 years.102
There are no studies to compare the biology of LCH in adults and children. The association of adult pulmonary LCH with smoking and evidence that the incidence of BRAFV600E mutations in adult LCH tissue is not the same as in the pediatric population indicates some differences. The LCs in adult lung lesions are mature DCs expressing high levels of the accessory molecules CD80 and CD86, unlike LCs found in other lung disorders.103 Molecular studies have shown pulmonary LCH in adults is primarily a reactive process, rather than a clonal proliferation as seen in childhood LCH.104 Subsequent investigations by this group with the Ion AmpliSeq technology showed two of five adult pulmonary LCH patients had the BRAFV600E mutation.105 An analysis of BRAFV600E expression by immunohistochemistry (IHC) and molecular techniques (allele-specific polymerase chain reaction [PCR] and Sanger sequencing) has also been reported for a series of adults with isolated pulmonary LCH and others with nonpulmonary lesions.106 Of the pulmonary LCH cases 7 of 25 (28 percent) were positive for BRAFV600E expression by IHC. The cumulative pack-years of smoking was significantly higher in the BRAF-positive adult pulmonary LCH patients than in the wild-type BRAF cases. Only 19 of 54 (35.2 percent) of the nonpulmonary cases had the BRAF mutation. The frequency of BRAFV600E mutation in North American pediatric series ranged from 57 to 65 percent based on deep sequencing and quantitative PCR.5,19 It is possible that technical differences in sensitivity underlie the relatively decreased reported frequency of BRAFV600E in adult cases of LCH. Further studies in adult patients will be needed to determine if age or ethnicity influence the role of BRAF mutations in LCH pathogenesis.
Adult LCH patients may have symptoms and signs for many months before a definitive diagnosis is made and treatment instituted. LCH in adults is often similar to that in children, except that isolated adult pulmonary LCH is closely associated with smoking. Presenting symptoms are (in order of decreasing frequency) dyspnea or tachypnea, polydipsia and polyuria, bone pain, lymphadenopathy, weight loss, fever, gingival hypertrophy, ataxia, and memory problems. Among the signs of LCH are skin rash, scalp nodules, soft-tissue swelling near bone lesions, lymphadenopathy, gingival hypertrophy, hepatosplenomegaly. Patients who present with isolated DI should be carefully observed for onset of other symptoms or signs characteristic of LCH. At least 80 percent of patients with DI had involvement of other organ systems: bone (68 percent), skin (57 percent), lung (39 percent), and lymph nodes (18 percent).107
Many patients have a papular rash with brown, red, or crusted areas ranging in size from a pinhead to a dime. In the scalp the rash is similar to seborrhea. Skin in the inguinal region, genitalia, or around the anus may have open ulcers that do not heal after antibacterial or antifungal therapy. In the mouth, swollen gums or ulcers along the cheeks, roof of the mouth, or tongue occur. In a series of 18 patients with skin LCH collected from 5 centers in the Netherlands followup revealed 5 developed malignancies which included 2 with myelomocytic leukemia, 1 with histiocytic sarcoma, and 2 with lymphomas. A literature review produced 6 additional cases of adults who had skin LCH and subsequently developed hematologic malignancies.108
The sites of bone involvement in adults differ from that of children. Lesions in the mandible occur in 30 percent of adults versus in 7 percent of children, and skull lesions in occur 21 percent of adults versus in 40 percent of children.102,109
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


