EBV is one of eight known human herpes viruses (HHVs) and is subgrouped into the
γ-herpes virus subfamily. Immature virus particles that measure 75 to 80 nm can be found in both the nucleus and the cytoplasm of infected cells. Mature, fully infectious particles, with a diameter of 150 to 200 nm, are found only in the cytoplasm.
12 The infectious virus particle has three components: a nucleoid, a capsid, and an envelope. The doughnutshaped central core, or nucleoid, contains the viral DNA in linear form. Surrounding the nucleoid is the capsid, which is icosahedral and is made up of hollow, tubular protein subunits called
capsomeres. Finally, the nucleocapsid (the capsid and the contained viral DNA) is enclosed in a protective envelope that is derived either from the nuclear membrane or from the outer membrane of the host cell. The envelope contains a number of viral antigens that are manufactured and inserted into the host cell membrane before the assembly of mature virus particles. The EBV genome (
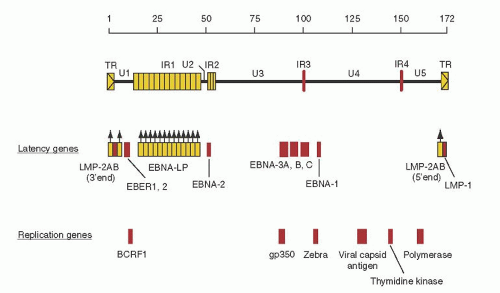
Fig. 62.1) is a double-stranded DNA molecule of approximately 173,000 base pairs (bp) organized into a series of unique coding regions (U1 to U5), divided by interval repeat regions (IR1 to IR4)
and terminal repeat domains. The genome encodes approximately 100 proteins.
13,
14 Figures 62.1 and
62.2 illustrate viral genes encoded by the genome and are discussed in detail in the section, “Epstein-Barr Virus Infection.” When EBV infects a cell, its linear genome circularizes to form an episome, an extrachromosomal element in the nucleus of the cell.
13,
15,
16,
17Many variants have been reported, although the relevance to human disease pathogenesis is unclear. Two variants EBV-1 (A type) and EBV-2 (B type) exist, which are differentiated mainly by their differences in the EBV-determined nuclear antigens (EBNAs) 2, 3A, and 3C.
13,
15,
18 Both types of EBV are found in most populations. The relationship between these different strains and specific diseases remains to be determined. Additional EBV variants with differences in EBNA-1 have been reported to be associated with specific EBV-affiliated diseases, such as BL, but the incidence of these variants has been shown to be similar in the general population of a particular geographic region.
19 Furthermore, mutations (so-called 30-bp or 69-bp deletions) of latent membrane protein (LMP-1) have been reported to be highly associated with the development of HL, certain forms of T- and B-cell lymphoma, and NPC.
20,
21,
22,
23 But again, when compared with EBV variants found in the geographic regions, there exists no difference between healthy individuals and those associated with tumors.
23,
24 Therefore, caution must be exercised when attributing specific EBV viral strains to particular diseases in the absence of geographically matched control isolates.
Epstein-Barr Virus Infection
The only host that the virus infects naturally is the human, although lymphocytes from other species, namely New World primates (i.e., cottontop marmosets, owl monkeys, and tamarins), are infectible.
25,
26 EBV has been found in a variety of human tissue types associated with disease (i.e., T lymphocytes, NK cells, HL, leiomyosarcoma, gastric carcinoma, hepatocellular carcinoma, and breast cancer).
27,
28,
29,
30,
31,
32,
33 The mechanism of EBV infection in these tumors remains to be fully elucidated.
Infection of B lymphocytes begins with attachment of the virus envelope glycoprotein (gp) 350/220 to the complement receptor C3d, also known as
CR2 or
CD21.
34 For entry into B lymphocytes, a complex of three viral proteins (gH-gL-gp42) is required, and human leukocyte antigen (HLA) class II molecules serve as a coreceptor for gp42.
35 Epithelium of the oropharynx is a source of viral replication,
36 but it has been difficult to determine the mechanism of infection, inasmuch as these cells do not express CD21 or HLA class II molecules. Recent studies have demonstrated that during primary infection, the majority of virions are not internalized after binding to B lymphocytes of the oropharyngeal tissue. And these B lymphocytes can form cellular conjugates with epithelial cells that allow for transfer of EBV virions into the epithelial cells.
37 The mechanism for EBV entry into other cell types (e.g., T cells, gastric mucosa, smooth muscle cells, etc.) may also be via direct contact with B cells that have bound virions with subsequent virion internalization. Infection of epithelium leads to the replication of the virus with the release of large quantities of viral particles containing three-part complexes (gH-gL-gp42) that favor infection of HLA class II and CD21-expressing cells (i.e., B lymphocytes located in the oropharyngeal lymphoid tissues of the Waldeyer ring).
35 These B lymphocytes may remain latently infected in the oropharynx, but if viral reactivation and replication occur, cellular lysis and death result, and the virus can be shed into the saliva. Virus contained in infectious saliva is produced by B lymphocytes of the oropharynx.
38,
39 Salivary virus may then be transmitted to another host.
The mechanisms underlying viral replication are not completely understood as there exist no in vitro models that recapitulate viral replication in humans. Viral replication is initiated through
oriLyt.
40 Two key early immediate genes are first transcribed, BRLF1 and BZLF-1, encoding Z EBV replication activator (ZEBRA) protein.
13,
15,
41 These proteins then up-regulate the expression of early gene products essential for viral replication, including viral DNA polymerase and viral thymidine kinase. Late gene products follow, including viral capsid antigen (VCA), the major envelope glycoprotein (gp350), and the viral protein BCRF-1, which contains 70% homology to interleukin (IL)-10.
13,
15,
42,
43 Complete viral replication results in the lysis and death of the host cell. Recently, it has been shown that viral proteins associated with type III latency (see below) are associated with viral replication.
44 What role this plays in B-cell transformation and immortalization remains to be determined.
Alternatively, infected B lymphocytes appear to undergo normal B-cell differentiation and disseminate throughout the body into secondary lymphatic organs (i.e., lymph nodes, spleen, and bone marrow).
44,
45,
46,
47 Studies have demonstrated that the reservoir for latent EBV infection is a small percentage of postgerminal center, antigen-selected resting memory B lymphocytes.
47 The number of latently infected B cells is approximately 10-5 to 10-6 of all B cells and remains stable for the majority of the life of the infected individual.
46,
48Latent infection is characterized by the expression of nine virally encoded proteins.
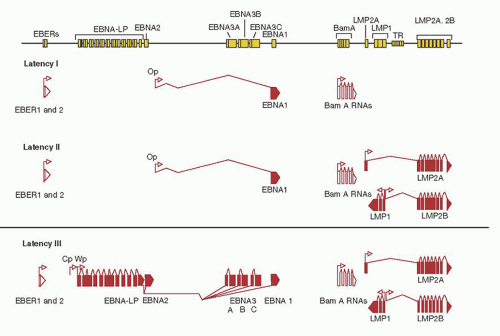
Figures 62.1 and
62.2 illustrate the transcriptions of the nine latent proteins of EBV (EBNA-1, EBNA-2, EBNA-3A, EBNA-3B, EBNA-3C, EBNA leader protein [LP], LMP-1, LMP-2A, and LMP-2B).
13,
15,
49 Two EBV-encoded RNAs (EBER-1 and EBER-2) are expressed abundantly in all EBV-infected cells and are valuable for making the diagnosis of EBV disease, but do not code for proteins and their function remains to be determined.
13 In addition, the Bam HI-A rightward transcript (BART) is generally found in infected cells, but its function also remains to be fully delineated.
50The state of latent infection is maintained by EBNA-1. The function of EBNA-1 is to bind to a nucleotide sequence called
ori-P, part of the DNA origin of EBV replication, thereby allowing the viral genome to be maintained in the nucleus of the latently infected B cell.
51,
52 By this mechanism, the EBV DNA is assured of being maintained in B cells that undergo replication.
EBNA-2 and LMP-1 appear to be required for B-cell immortalization.
13 EBNA-2 transactivates the expression of LMP-1 and LMP-2.
13,
53 EBNA-LP augments the ability of EBNA-2 to up-regulate LMP-1. LMP-1 and LMP-2 are associated with a cellular tyrosine kinase. The function of LMP-1 is similar to that of other members of the tumor necrosis factor (TNF) receptor family and is similar—but not identical—to that of CD40. LMP-1 interacts with TNF-receptor-associated factors and the TNF-receptor-associated death domain in infected cells and activates nuclear factor-
kB and c-Jun N-terminal kinase pathways, resulting in B-cell activation and proliferation.
49,
54,
55,
56,
57 Additionally, LMP-1 expression also induces bcl-2 expression and can prevent apoptosis in B cells.
57 LMP-2 prevents viral reactivation by blocking tyrosine phosphorylation and promotes B-cell survival.
58 The EBNA-3 proteins regulate expression of certain cellular genes, including specific cellular receptors such as CD28, CD19, CD21, CD23, and CD30; T-cell co-stimulatory molecules CD80/CD86; adhesion molecules such as intercellular adhesion molecule-1, leukocyte factor antigen-1, and leukocyte factor antigen-3, and a member of the src oncogene family, c-fgr.
15,
59Expression of EBV genes varies along the spectrum of EBV-associated diseases and often differs from in vitro immortalized B cells or normal human resting B cells infected by EBV.
49,
53 The viral gene expression of viral latency is illustrated in
Figure 62.2. Briefly, in type I latency- EBNA-1, EBER-1, and EBER-2 and BART are expressed. Type I latency is observed in BL and a portion of EBV-positive gastric carcinoma. Type II latency is characterized by the expression of EBNA-1, LMP-1, LMP-2A and B, EBER-1, and EBER-2 and is observed in NPC, and other EBV-positive lymphomas, such as T- or NK-cell NHL, and the Reed-Sternberg (RS) cells of HL. EBV-infected cells of LPD observed in immunodeficient patients resemble in vitro immortalized B cells and generally express all nine of the EBV-related latent proteins (type III latency). The site of EBV latency in seropositive healthy individuals, the resting memory B cells does not appear to express EBNA-1 but only LMP-2, EBER-1, and EBER-2 together with BART (type IV latency).
60
Immune Response to Epstein-Barr Virus Infection
Understanding the immune response to EBV infection is essential to comprehend the pathogenesis of EBV-related disease. EBV is a very potent immune stimulus. The immune system controls lymphoproliferation in the normal host and maintains a host/virus symbiosis.
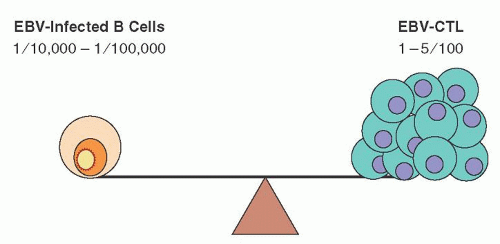
Figure 62.3 illustrates the delicate balance between the host T-cell immune response and control of B-cell proliferation of latently infected B cells. In a healthy individual, although only 10
-5 to 10
-6 B cells are latently infected with EBV, approximately 1% to 5% of all circulating CD8
+ T cells are capable of reacting against EBV.
48,
61,
62,
63 Initially, there is B-cell proliferation, producing both EBV-specific and -nonspecific antibodies. The number of these virus-containing B cells rises during the acute phase of the infection but never exceeds 0.03% to 0.1% of the circulating mononuclear cells.
64,
65 A cellular immune response follows, which is composed primarily of an expansion of cytotoxic T lymphocytes (CTLs), with the vast majority being EBV-specific, although some are not EBV-specific.
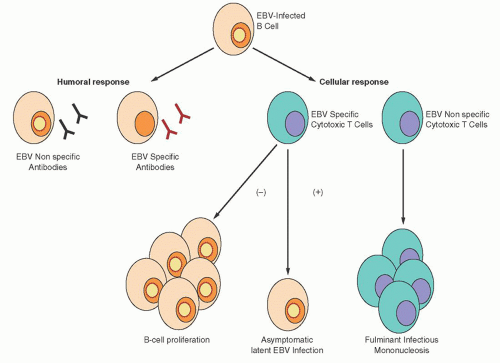
66,
67 Figure 62.4 illustrates the pathologic consequences of an abnormal CTL response to EBV infection. A deficient CTL response, either quantitative or qualitative, results in an EBV-driven B-cell proliferative process. The lack of an appropriate CTL response can also result in an aggressive, predominantly T-cell and histiocytic reaction that is not EBV-specific. This reaction is characterized by extensive infiltration of lymphoid and parenchymal organs with hemophagocytosis, and tissue destruction is often observed. If unchecked, this reaction can be rapidly fatal.
68,
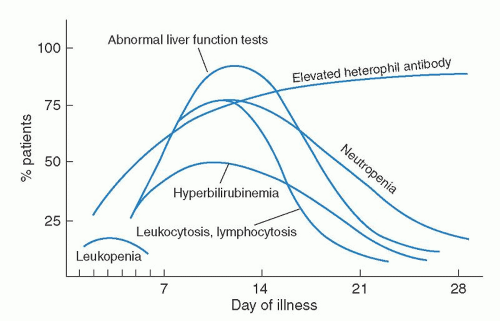
69The humoral response to EBV is well characterized (
Fig. 62.5).
70,
71,
72,
73 VCA antibodies are the earliest to appear: first immunoglobulin M (IgM) and, later, IgG. IgM antibodies probably arise during the incubation period, peak with symptoms, and then decline rapidly.
73 IgA anti-VCA antibodies are seen in some patients and, like IgM anti-VCA antibodies, are gone within several weeks.
74,
75 IgG anti-VCA antibodies reach a peak 2 to 3 weeks after their IgM predecessors and persist for life.
74,
75 The majority of patients also have a transient response to the EBV early antigen (EA), peaking usually within a month of infection.
72 Antibodies to EBNA may appear several weeks after the onset of the illness in some patients but, in general, take several months to appear, and titers rise slowly over 1 to 2 years and persist for life. The majority of normal individuals have detectable IgG to EBNA by 6 months after EBV infections, although it may take years to develop detectable anti-EBNA titers.
76 In young children, the anti-VCA and anti-EA responses may be much less intense, and anti-EBNA may take much longer to be seen.
77An EBV-nonspecific humoral response also occurs with EBV infection. Paul et al.
78 first described this phenomenon in 1932 when they reported that the sera of patients with IM contained heterophil antibodies against sheep erythrocytes in concentrations far above normal. The use of heterophil antibodies in the diagnosis of IM is discussed in detail in the section, “Infectious Mononucleosis: Diagnosis.” Many of these EBV-nonspecific antibodies function as autoantibodies and include cold reactive anti-i antibodies, Donath-Landsteiner cold hemolysins, and antibodies against smooth muscle, thyroid, and stomach.
79,
80,
81,
82 In addition, antinuclear antibodies have been found in the sera of some patients,
83,
84 as well as rheumatoid factors,
85 including anti-Gm antibodies,
86 anticardiolipin antibodies, and antiactin and anticytoskeletal antibodies.
87 The serum of some persons with IM may contain cryoglobulins.
88 Some of these autoantibodies have been reported to be monoclonal.
84Although neutralizing antibodies produced after primary infection may play a role in thwarting the spread to additional B cells, EBV-specific antibodies are most useful for diagnosis,
whereas the cellular response is the most important for control of EBV infections. NK cells and CD4
+ T cells have been shown to play a role, but CD8
+ memory cytotoxic T cells (EBV-CTLs) are the primary defense in controlling EBV infections.
66,
67,
89,
90 The absolute number of NK cells may initially be increased, but a decrease in NK function is usually observed, returning to normal gradually over several weeks.
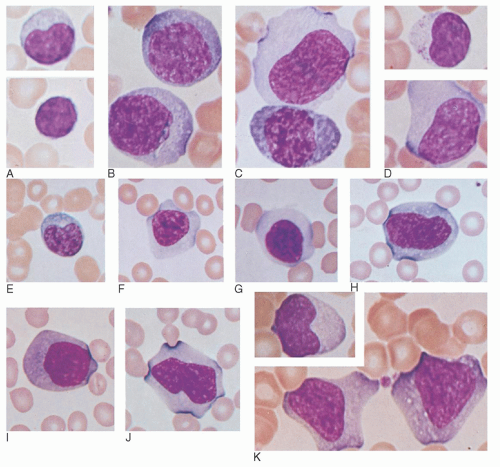
91 Initially, these EBV-CTLs account for the majority of cells causing the lymphocytosis and the large, pleomorphic, atypical lymphocytes, or “Downey cells,” characteristic of IM.
66,
67,
90,
92 Of note, it has been shown that the amount of these T cells in the peripheral blood correlates with symptoms seen in IM, not the number of EBV-infected B cells or the viral load.
93 Not surprisingly, at first the majority of EBV-CTLs are reactive against lytic viral antigens. During lytic infection three EBV proteins, BNLF2A, BILF1, and BGLF5, function to lower HLA I surface expression, which in effect helps EBV-infected cells evade the immune system.
94 However, over time the number of EBV-CTLs reactive against latent viral antigens predominates.
66,
67This symbiosis of EBV and the infected host is maintained by interactions between viral gene expression in latently infected B cells and host EBV-CTL surveillance. The reservoir of viral latency is found among the resting memory B cells.
46,
47 These resting memory B cells do not express high levels of adhesion molecules or T-cell co-stimulatory molecules, making them poor antigen-presenting cells. When these cells divide, EBNA-1 is expressed to ensure passage of viral DNA to progeny cells.
52 Although CD4
+ T-cell responses against EBNA-1 have been documented,
95 EBNA-1-specific cytotoxic T cells are not readily generated.
96,
97 Thus, the reservoir of latent EBV infection is not recognized readily by the immune system.
Activated EBV-infected B cells express major histocompatibility complex class I antigens; various adhesion molecules make these proliferating EBV-infected B cells good antigen-presenting cells. Proliferating, latently infected B cells express all latent proteins, including EBNA-2, EBNA-3A, EBNA-3C, LMP-1, and LMP-2 (type III latency). These proliferating EBV-infected B cells are highly susceptible to cellular lysis by EBV-CTL and are eliminated during convalescence, whereas memory EBV-CTL provide lifetime immunosurveillance against EBV-driven B-cell proliferation. The immunodominant epitope of the EBV-CTL response is highly restricted by the HLA type of the individual.
98 For months after the expansion of EBV-infected B cells is controlled, virus is shed at high titers in the saliva,
99 indicated ongoing viral replication in infected B cells of the oropharanyx.
46 Recent studies have demonstrated that EBV-CTL against latent viral antigens home more efficiently to lymphoid tissue of the oropharynx than EBV-CTL against lytic viral antigens.
100 This observation helps explain that the greatest risk of acquiring primary infection is salivary exposure from a recently infected person or a person in early convalescence, although infective virions can be found in the saliva of persons with a long latent infection.