Immunodeficiency Disorders and Neoplasias of the Lymphoid System
Introduction
At first glance, the connection between immunodeficiency syndromes and neoplasias of the lymphoid system is not apparent: Immunodeficiency syndromes are characterized by absences or deficiencies, whereas neoplasia reflects excesses or uncontrolled proliferations. So why are they discussed in the same chapter? The relationship between these two types of immune system disorders demonstrates the fine tuning and integration of the elements of the immune system. Deficiencies, particularly in a single arm of the immune system, affect the ability of the remaining elements to control their growth. For this reason, immunodeficiencies are fertile ground for the development of neoplasia. Because autoimmune phenomena are further manifestations of the loss of immune regulation that frequently accompanies immunodeficiency, three seemingly disparate disease states—immunodeficiency, autoimmunity, and lymphoid neoplasia—often coexist in a single individual. Primary autoimmune diseases were discussed in Chapter 13; autoimmune reactions resulting from immunodeficiency or leading to lymphoid malignancy will be highlighted throughout this chapter.
As you have learned in previous chapters, the immune response is mediated by T and B lymphocytes, natural killer (NK) cells, myeloid/monocytic lineage cells, dendritic cells, and complement. The interactions among these cells, their soluble mediators (antibodies and cytokines), and complement are tightly controlled. Disorders in the development and differentiation of the cells, synthesis of their products, or interactions among them may lead to immune deficiencies with clinical manifestations ranging from mild to fatal in severity. However, clinical consequences of deficiencies in which the immune system shows redundancy (overlap in the functions of one component with another) are noticeably absent. For example, parts of the cytokine network normally exhibit redundancy and rarely come to medical attention.
Although inborn immunodeficiency diseases (conditions present at birth) are generally rare, early descriptions of these “experiments of nature” shed light on the functioning of the immune system. Animal models that mimicked different types of human immunodeficiencies illuminated the cellular subdivisions of adaptive immunity into T and B lymphocytes—that is, cell mediated versus humoral immunity. In addition, elucidation of the genetic basis of multiorgan autoimmune syndromes (e.g., mutations or deficiencies in molecules expressed predominantly in the thymus), has led to a better understanding of immune regulation and central tolerance.
Today, information gained from analyzing these rare immunodeficiency syndromes and lymphoid neoplasms at the molecular level is applied to their treatment and to the development of immunotherapies for autoimmune diseases and lymphoid and nonlymphoid malignancies. The chapter begins with descriptions of inborn and acquired immunodeficiency syndromes and concludes with neoplasias of the immune system.
Immunodeficiency Syndromes
Immunodeficiencies are divided into two major categories: primary immunodeficiencies, which may be hereditary or acquired, in which the deficiency is the cause of disease; and secondary immunodeficiencies, in which the immune deficiency is a result of other diseases or conditions.
We will first concentrate on those primary immunodeficiencies whose manifestations result in an increase in susceptibility to infection. These can be categorized on the basis of clinical presentation or on the basis of the arm of the immune system that is malfunctioning. These two classification schemes roughly correspond to each other and divide as follows: (1) T- or cell-mediated immunity; (2) B- or humoral (antibody-mediated) immunity; (3) both B- and T-cell immunity; (4) innate immunity mediated by phagocytic cells and/or NK cells; and (5) complement activation. Abnormalities of cytokines, chemokines, and their receptors do not form a separate category but are incorporated into groups 1–4 since they are the means by which these cells communicate and function.
An expressed immune response is often the result of interactions among several cell types; for example, a deficiency of antibody production and B-cell function may actually be caused by an underlying problem in T cells or in the T–B-cell interaction. Classification based on the apparent expressed defect, rather than its underlying cause (which may be unknown), is a useful framework for diagnosing new patients. This method of classification also allows correlation with animal models in which the fundamental immune defect may be more readily identified.
Immunodeficiency should always be suspected in a patient with recurrent infections. As shown in Table 18.1, the types of infections can often facilitate diagnosis of the underlying problem. For example, recurrent bacterial otitis media (ear infection) and bacterial pneumonia are common in individuals with B-cell and antibody deficiencies. Increased susceptibility to fungal, protozoal, and viral infections is seen with T-cell and cell-mediated immunodeficiencies. Systemic infections with bacteria that are normally of low virulence, superficial skin infections, or infections with pyogenic (pus-producing) organisms suggest deficiencies in phagocytic cells. Recurrent infections with pyogenic microorganisms are associated with complement deficiencies. Of particular significance is the occurrence of opportunistic infections, diseases caused by microorganisms present in the environment that are nonpathogenic in immunocompetent individuals. Pneumocystis jiroveci (P. jiroveci), cytomegalovirus (CMV), Toxoplasma gondii, Mycobacterium avium, and Candida are among the most common culprits causing opportunistic infections in patients with deficiencies in cell-mediated immunity.
Table 18.1. Major Clinical Manifestations of Immune Disorders
| Disorder | Associated Diseases |
|---|---|
| Deficiency | |
| B-lymphocyte deficiency: deficiency in humoral or antibody-mediated immunity | Recurrent bacterial infections such as otitis media and recurrent pneumonia |
| T-lymphocyte deficiency: deficiency in cell-mediated immunity | Increased susceptibility to viral, fungal, and protozoal infections |
| T- and B-lymphocyte deficiency: combined deficiency of antibody and cell-mediated immunity | Acute and chronic infections with viral, bacterial, fungal, and protozoal organisms |
| Phagocytic cell deficiency | Systemic infections with bacteria of usually low virulence; infections with pyogenic bacteria; impaired pus formation and wound healing |
| NK cell deficiency | Viral infections, associated with several T-cell disorders and X-linked lymphoproliferative syndromes |
| Complement component deficiency | Bacterial infections; autoimmunity |
| Unregulated Excess | |
| B lymphocytes | Monoclonal gammopathies; other B-cell malignancies; lymphoproliferative syndromes |
| T lymphocytes | T-cell malignancies; autoimmune lymphoproliferations, autoimmune syndromes |
| Complement components | Angioedema due to defect in Cl esterase inhibitor |
The timing of infections is also a hint to the underlying diagnosis. Infants are protected for the first 6 months to 1 year of life by maternal antibodies transferred through the placenta and breast milk. Therefore deficiencies in humoral immunity exclusively are manifest beginning at approximately 6 months to 1 year of age. In contrast, defects in T-cell–mediated immunity or combined defects in T- and B-cell function often present from birth as failure to thrive.
Primary Immunodeficiency Syndromes
With the exception of IgA deficiency, the frequency of primary immunodeficiency syndromes is very low—about 1 in 10,000. Approximately 50% of all cases are antibody deficiencies, 20% are combined deficiencies in antibody- and cell-mediated immunity, 18% are phagocytic disorders, 10% are disorders of cell-mediated immunity alone, and 2% are complement deficiencies. Figure 18.1 shows that, in general, the earlier the genetic defect or block occurs in development, the more arms of the immune system are affected and the more severe the disease.
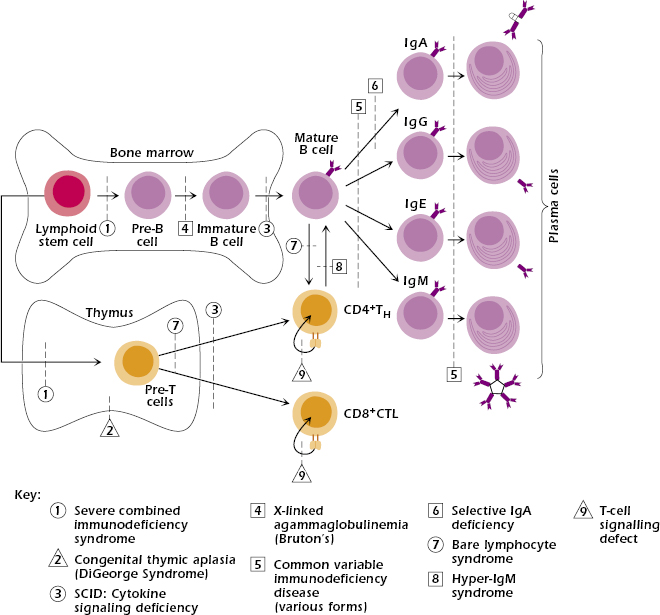
Severe Combined Immunodeficiency Diseases (SCID).
SCID comprises a heterogeneous group of diseases in which both cell-mediated immunity and antibody production are defective (see Figure 18.1). Individuals with SCID are susceptible to virtually every type of microbial infection (viral, bacterial, fungal, and protozoal), including opportunistic infections, most notably CMV, P. jiroveci, and Candida. Vaccination with attenuated live virus could prove fatal in infants with SCID.
Patients can be subclassified at initial evaluation according to the lymphocyte subsets present in their blood (Table 18.2). One group, designated as T−B+, has essentially no T cells with normal or increased numbers of nonfunctioning B cells. This group of patients may also lack NK cells. A second group, T−B−, has severe lymphopenia due to the absence of both T and B cells. A few patients are T+B+, and rare patients are T+B−. The preferred treatment for all SCID patients is a T-cell-depleted bone marrow transplant from an HLA-matched sibling donor.
Table 18.2. Severe Combined Immunodeficiency Diseases
| Specific Disorder | Underlying Deficiency | Mode of Inheritancea |
|---|---|---|
| T−B+ subgroup | ||
| X-linked SCID | Mutated γ chain of cytokine receptors | X linked |
| Autosomal recessive SCID | Mutated JAK3 tyrosine kinase | AR |
| T−B− subgroup | ||
| Adenosine deaminase deficiency | ADA enzyme | AR |
| Purine nucleoside phosphorylase deficiency | PNP enzyme | AR |
| Recombinase deficiency | Rag 1 or Rag 2 enzyme | AR |
| T+B− Subgroup | ||
| Omenn syndrome | Partial Rag deficiency (most common) | AR |
| T+B+ Subgroup | ||
| Bare lymphocyte syndrome | MHC class II transcription activator (4 proteins) | AR |
| MHC class I TAP defect | AR | |
| ZAP-70 deficiency | Kinase domain of TCR-associated PTK, ZAP-70 | AR |
| Multisystem Disorders | ||
| Wiskott–Aldrich syndrome | WAS protein | X linked |
| Ataxia telangectasia | ATM protein for DNA repair | AR |
aAR, autosomal recessive.
T−B+ Subgroup.
X-Linked SCID. Patients with X-linked SCID constitute 40–50% of SCID cases, with the majority of those showing T−B+ lymphopenia and lacking NK cells. Mutations have been found in the gene located on the X chromosome that codes for the γ chain, a chain common to the receptors for interleukin-2 (IL-2), IL-4, IL-7, IL-9, and IL-15 (see Chapter 12). Thus, the mutation impairs responses to a multitude of cytokines (Figure 18.2A).
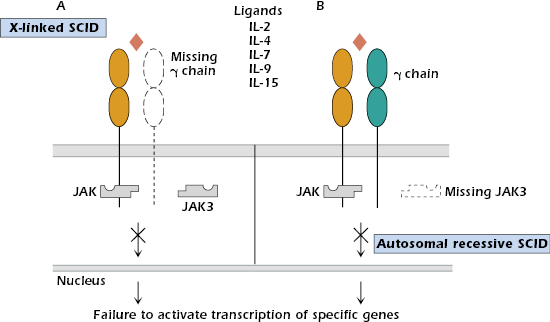
Autosomal Recessive SCID. A small subgroup of patients characterized by T−B+ NK− lymphopenia shows an autosomal recessive (rather than X-linked) pattern of inheritance. These individuals have a phenotype identical to the X-linked SCID group and cannot be distinguished clinically. Mutations are localized in the gene for JAK3 tyrosine kinase (Figure 18.2B), the intracellular molecule responsible for transmitting signals from the γ chain of the cytokine receptors (see Chapter 12). Expression of JAK3 is normally restricted to hematopoietic cells.
An even smaller group of patients with autosomal recessive SCID present with T−B+ NK+ lymphopenia. These individuals have mutations in the IL-7Rα chain or in a chain of CD3.
Animal models with targeted defects have proven very instructive in delineating these human deficiencies. In mouse gene knockout models (see Chapter 6), γ-chain knockouts, like SCID patients, have defective development of both T- and B-cell lineages. IL-7 and IL-7R knockouts bear a greater resemblance to SCID patients, suggesting that, in the mouse, IL-7 is crucial for T-cell and B-cell development and/or function and that the absence of IL-7 is not compensated for by other cytokines. In contrast, IL-2 knockouts show only some immune dysfunction, with normal T- and B-cell development and without a SCID phenotype.
T+B− Subgroup.
Adenosine Deaminase Deficiency. Adenosine deaminase (ADA), an enzyme in the purine salvage pathway, is a ubiquitously expressed housekeeping enzyme (an enzyme used in the everyday function of all or most cells). Individuals lacking this enzyme account for approximately 20% of SCID patients and show an autosomal recessive pattern of inheritance. The deficiency results in buildup of toxic wastes, causing symptom progression over time and making early detection and treatment particularly critical in these patients.
ADA deficiency has its greatest impact on the immune system, resulting in failure of both T- and B-lymphocyte development. Many patients have an associated characteristic skeletal abnormality. The reason these patients do not exhibit even more multisystem problems is not completely understood. Investigation of this rare genetic disease has shown the particular importance of the salvage pathway in lymphocyte development and differentiation, and has led to the development of antileukemic drugs to stop the growth of malignant lymphocyte precursors.
ADA-deficient patients lacking a matched sibling marrow donor were the first group to be treated with gene therapy by transfecting a functional gene for ADA using a retroviral vector. Although gene therapy has been most successful in the small number of ADA-deficient patients it has been tried on (approximately 40 to date), even after years of development, gene therapy as an experimental approach for treating genetic disorders remains fraught with great difficulties. Interestingly the ADA-deficient patients are the only group that seems free from the subsequent development of a T-cell leukemia, suggesting that the ADA-defective cells cannot support transformation. Continuous enzyme supplementation is an alternative treatment for these patients.
Purine Nucleoside Phosphorylase Deficiency. A mutation in another enzyme in the purine salvage pathway, purine nucleoside phosphorylase (PNP), also leads to a buildup of toxic products that are particularly damaging to the neurologic system and to T cells. Eventually, all lymphoid tissues—thymus, tonsils, lymph nodes, and spleen—are depleted. Paradoxically, even though children with this condition are markedly immunodeficient, autoimmune disease is common in these patients.
Recombinase Deficiencies and Radiosensitivity SCID. Recombination-activating genes (RAG) 1 and 2 code for enzymes involved in the rearrangement of the Ig genes in pre-B cells and the TCR genes in pre-T cells (see Chapters 7 and 10). Both enzymes are absolutely required for gene rearrangement, so mutations in either result in complete absence of T cells and B cells. Maturation stops at the pre-T- and pre-B-cell stages. Typically, NK-cell function is intact. Mutations in genes for other proteins (such as Artemis), which are involved in Ig and T-cell receptor (TCR) recombination and DNA repair, will generate a similar clinical picture. The relationship with DNA repair accounts for the radiosensitivity.
Omenn Syndrome. Omenn syndrome refers to a clinical presentation in which the infants have severe immunodeficiency to every type of pathogen, along with enlarged lymphoid tissues (lymphadenopathy), high IgE levels, high eosinophil levels in the peripheral blood and severe erythroderma. Many of clinical parameters are similar to those seen in severe graft-versus-host (GVH) disease (discussed in Chapter 19). Omenn syndrome patients demonstrate dysregulation of the immune system resulting in an attack against self. These patients are T+B− by peripheral blood analysis and have massive skin and gastrointestinal infiltration by eosinophils and activated T cells, which produce TH2-type cytokines (see Chapters 11 and 12). This results in the hyper-IgE syndrome and malnutrition due to protein loss. The majority of Omenn syndrome patients have reduced but partial Rag activity (leaky SCID). Patients with this clinical picture may alternatively have mutations in other molecules commonly associated with SCID, such as ADA and Artemis (a DNA repair protein) or may have mutations in an RNA component of mitochondrial RNA processing endoribonuclease. The success rate of bone marrow transplants in individuals with Omenn syndrome is low compared with other types of SCID patients; failures are due to graft rejection. Thus, although Omenn syndrome patients are immunodeficient, they require pretreatment with immunosuppressive therapy.
T+B+ Subgroup.
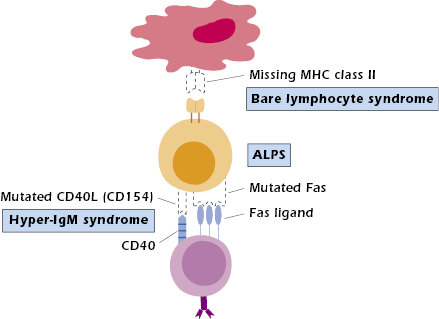
Bare Lymphocyte Syndrome. Bare lymphocyte syndrome (BLS) results from the failure to express HLA (the human major histocompatibility complex [MHC]) molecules and is thus a defect in antigen presentation rather than an intrinsic lymphocyte cellular abnormality. BLS is divided into three groups, depending on which class of HLA molecules is missing: class I, class II, or both classes I and II. Only those individuals lacking expression of HLA class II molecules consistently show immunodeficiencies. Circulating T- and B-cell numbers may be normal; however, in the absence of HLA class II molecules, protein antigens cannot be presented to CD4+ T cells (Figure 18.3). Therefore, collaboration does not occur between antigen-presenting cells (APCs; B cells, macrophages/monocytes, dendritic cells) and CD4+ T cells. As a result, help is not provided to B cells for antibody production or to T cells for cytotoxic T-cell generation (see Chapter 9). This results in a clinical presentation of combined immunodeficiency. Since MHC class II expression is required on thymic epithelial cells for positive selection of CD4+ T cells, proportionately fewer CD4+ T cells are produced in the thymus (see Figure 18.1, defect 7). Therefore, most patients have a decreased proportion of CD4+ to CD8+ T cells, resulting in a reversed CD4:CD8 T-cell ratio. Any CD4+ T cells present are functional, as demonstrated by their ability to respond when stimulated in vitro. Since GVH disease can still occur in these patients, HLA matched bone marrow donors are required for treatment.
The mutation responsible for BLS affecting class II molecules is not in the HLA class II genes themselves but in one of the four genes that code for regulatory factors required to transcribe the class II genes. A better understanding of this transcription failure in BLS could result in the development of methods to turn off HLA class II expression. Theoretically these could then be applied to prevent graft rejection of transplanted organs (such as the kidney and liver) in immunocompetent individuals.
Few patients deficient in HLA class I expression have been identified; some were discovered serendipitously. This is undoubtedly due to the fact that not all individuals with deficient HLA class I expression show clinically significant immunodeficiency. Those who do, usually present with chronic inflammatory lung disease late in childhood. As in BLS patients with defective HLA class II expression, patients with HLA class I deficiency do not have a mutation in the HLA class I gene, but rather have a mutated gene for the transporter protein (Tap). As described in Chapter 9, Tap transports peptides generated in the cytosol into the endoplasmic reticulum, where they interact with and stabilize the structure of MHC class I molecules. In the absence of the Tap gene product, expression of MHC class I molecules on the cell surface is very low. In the case of HLA class I deficiencies, positive selection of CD8+ T cells in the thymus is defective, and their peripheral blood levels are decreased. For reasons that are unclear, when patients with this defect are symptomatic, they have recurrent bacterial pneumonias rather than the expected viral infections.
ZAP-70 Mutation. Patients with a mutation in the T-cell tyrosine kinase ZAP-70, which transduces the signal transmitted through the TCR, also present with a SCID-like phenotype. Originally it was not apparent why patients defective in ZAP-70 expression would present with a SCID-type clinical picture. ZAP-70 plays a critical role in the function of mature T cells (see Figure 18.1, defect 3). In addition, although the peripheral blood counts, lymph nodes, and thymus are essentially normal, CD8+ T cells are missing in patients defective in ZAP-70 expression. This indicates that ZAP-70 is also required for CD8+ T-cell differentiation in the thymus. Recent evidence indicates that ZAP-70 is expressed in developing B cells and in some mature B cell subsets but at a low level. Its function is redundant with a homologous tyrosine kinase molecule, Syk. Thus the role of ZAP-70 in B-cell development and function remains unclear.
Other Multisystem Disorders.
In addition to the combined immunodeficiency diseases we have just discussed, several multisystem inherited disorders result in a SCID-like clinical picture.
Wiskott–Aldrich Syndrome. Wiskott–Aldrich syndrome is an X-linked disease showing a classic triad of symptoms: (1) bleeding diathesis (tendency to bleed) due to thrombocytopenia (low platelet level in blood) and small platelet size, (2) recurrent bacterial infections, and (3) paradoxically, allergic reactions (including eczema, elevated IgE levels, and food allergies). Over the longer term, patients have an increased risk of developing malignancies, particularly of the lymphoid system. The genetic basis of the disease is a mutation in the X-linked gene coding for the Wiskott–Aldrich syndrome protein (WASP), which is expressed in all hematopoietic stem cells. The WASP interacts with the cytoskeleton; it allows remodeling after receptor engagement, contributes to the immune synapse between interacting cells, and affects maturation and migration of cells of both the adaptive and innate immune systems. In cells of patients with this syndrome, the cytoskeleton cannot effectively reorganize in response to stimuli.
The immune defects are variable, but both T and B cells are functionally abnormal, with T-cell numbers particularly decreased. Characteristically, patients are unable to respond to polysaccharide antigens. Treatment consists of antibiotics and antiviral agents given promptly with each infection. Reconstitution of T and B cells has been reported following bone marrow transplantation. Without treatment, the average life expectancy is approximately 3 years. With extension of survival, the incidence of malignancies would be expected to increase.
Ataxia Telangiectasia. Ataxia telangiectasia (AT) is another multisystem genetic disorder in which neurologic symptoms (staggering gait or ataxia) and abnormal vascular dilatation (telangiectasia) accompany increased susceptibility to infections; lymphopenia (low lymphocyte numbers in peripheral blood); thymic hypoplasia; and depressed levels of IgA, IgE, and sometimes IgG. The immune defect involves both cellular and humoral (T-cell-dependent and T-cell-independent) immune responses; T-dependent regions of lymphoid tissues are affected most severely. The genetic basis of this syndrome is a mutation in the gene coding for the protein ATM, part of a pathway activated when the cell suffers DNA breaks from ionizing radiation and oxidative damage. AT patients have impaired development of T and B cells. The normal generation of both lineages involves critical phases of extensive cell proliferation, apoptosis, and DNA recombination events; all of these may be dysregulated with a mutated, nonfunctional ATM protein. AT children also have a greatly increased risk of developing malignancies, particularly lymphoid neoplasms. This may be the result of an ATM-dependent defect in DNA repair or cell cycle arrest following chromosomal damage. AT has been grouped with Bloom syndrome and Fanconi anemia; all three disorders show similar variable immunodeficiencies and susceptibility to DNA damage.
In summary, the causes underlying severe defects in both cell-mediated and humoral immunity are varied. They range from mutations in enzymes found in all cells, which should have global effects in the body (such as deficiencies in ADA and PNP), to mutations involving signaling proteins specifically expressed in T cells (such as the ZAP-70 mutation).
Animal models have been informative, both in understanding the defects observed in human syndromes and in helping delineate steps in normal T- and B-cell development. The SCID mutant mouse strain, which has a genetic defect in a protein repairing double-stranded DNA breaks, was the first mouse model used for the study of this group of diseases. Since then, knockout mouse models for the majority of these spontaneous human genetic diseases have been produced as a means to study these diseases (see Chapter 6). In addition, SCID mice and nude mice (discussed later in this chapter), with diminished ability to reject foreign tissues, can be used as “living test tubes” to study the growth of human hematopoietic stem cells and human tumors.
Immunodeficiency Disorders Associated with T Cells and Cell-Mediated Immunity
As noted in Table 18.1, patients with T-cell-associated deficiency diseases are susceptible to viral, fungal, and protozoal infections. In addition, because T cells are required to help B cells produce antibodies to T-dependent antigens (see Chapter 11), patients with T-cell-associated deficiencies also exhibit selective defects in antibody production. Consequently, T-cell-deficient patients may be difficult to distinguish clinically from SCID patients.
Congenital Thymic Aplasia (DiGeorge Syndrome).
DiGeorge syndrome is a T-cell deficiency in which the thymus, as well as several nonlymphoid organs, develops abnormally. The syndrome is caused by defective migration of fetal neural crest cells into the third and fourth pharyngeal pouches. This usually takes place during week 12 of gestation. In DiGeorge syndrome, the heart and face develop abnormally, and the thymus and parathyroids fail to form, resulting in thymic aplasia and hypoparathyroidism along with cardiac disease. Due to the lack of thymic structure, there is an absence of mature T cells, resulting in immunodeficiency. The athymic nude mouse is an animal model of DiGeorge syndrome; in these animals, the thymus and hair follicles do not develop.
DiGeorge syndrome is not hereditary; it occurs sporadically and is generally the result of a deletion in chromosome 22q11. Newborns present with hypocalcemia (low serum calcium levels) from absence of the parathyroid glands and have symptoms of congenital cardiac disease. Affected children suffer from recurrent or chronic infections with viruses, bacteria, fungi, and protozoa. They have either no or very few mature T cells in the periphery (blood, lymph nodes, or spleen) (see Figure 18.1, defect 2). Although B cells, plasma cells, and serum Ig levels may be normal, many patients fail to mount an antibody response after immunization with T-dependent antigens. The lack of helper T cells, which are required for Ig isotype switching, results in the absence of IgG and other switched isotypes following immunization. The IgM response to T-independent antigens is intact. Since individuals with DiGeorge syndrome lack T cells and fail to generate normal antibody responses, they should never be immunized with live attenuated viral vaccines.
In the past, children with DiGeorge syndrome were treated with a fetal thymus graft, which resulted in the appearance of host-derived T cells within a week. The fetal thymus used for transplantation needed to be less than 14 weeks’ gestation to avoid GVH reactions, which would occur if donor thymocytes were transferred into the immunoincompetent recipient. This donor fetal thymus provided the environment (the thymic epithelial cells) for development of recipient T cells from the patient’s normal lymphoid precursors. Although the T cells produced were normal, cell-mediated immunity and help for antibody production were not fully restored. The recipient’s T cells learned the MHC of the transplanted thymus as “self” and sometimes collaborated poorly with the body’s own APCs in the periphery (see Chapters 9 and 10). Because this treatment strategy was not entirely successful, therapy now is mostly in response to symptoms. Some patients have small remnants of thymic tissue, allowing delayed (though still diminished) T-cell maturation. The other medical problems associated with the syndrome, such as congenital heart disease, add to the overall poor prognosis.
T-Cell Deficiencies With Normal Peripheral T-Cell Numbers.
A number of patients have been identified with functional rather than numerical defects in their T cells. Clinically, they may present with opportunistic infections and a strikingly high incidence of autoimmune disease. Family studies show autosomal recessive patterns of inheritance. Molecular analysis demonstrates that the underlying causes are heterogeneous, with deficient expression of ZAP-70 tyrosine kinase, CD3ε, or CD3γ (Figure 18.4). As previously discussed, patients with ZAP-70 deficiencies present with SCID-like clinical phenotypes.
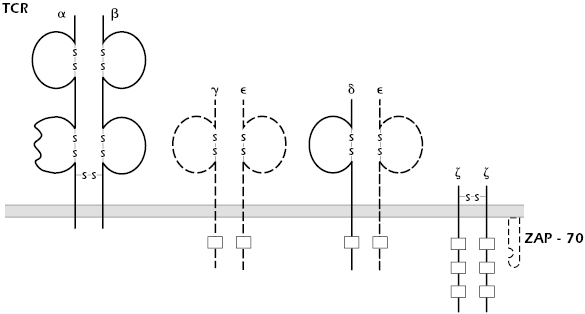
Mutations in CD3 chains are quite rare; only a handful of patients with such defects have been described. Mouse models confirm that all the CD3 peptide chains are required for normal signaling through the TCR. It is not clear, however, that these mouse models accurately mimic the few patients reported.
Chronic Mucocutaneous Candidiasis.
Chronic mucocutaneous candidiasis is a poorly defined collection of syndromes characterized by Candida infections of skin and mucous membranes. This ubiquitous fungal organism is normally nonpathogenic. These patients usually have normal T-cell–mediated immunity to microorganisms other than Candida and normal B-cell-mediated immunity (antibody production) to all microorganisms including Candida. Thus, they have only a selective defect in the functioning of T cells. This disorder affects both males and females, is particularly prevalent in children, and may be inherited.
Defects in Control.
Three syndromes, each the result of gene mutations, are characterized by lack of tolerance and/or overwhelming, multiorgan autoimmune diseases. One affects the transcription factor autoimmune regulator (AIRE), expressed on thymic epithelial cells and responsible for negative selection of T cells. Its resulting syndrome, autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) [also known as autoimmune polyendocrine syndrome type 1 (APS1)] is characterized by autoimmune disease in multiple endocrine organs. The second affects the transcription factor Foxp3 resulting in an absence of T-regulatory cells and the disease immunodysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX). In the last one of these syndromes, a surface molecule important in inducing cell death and thereby eliminating unwanted cells is mutated, resulting in autoimmune lymphoproliferative syndrome (ALPS). These syndromes were discussed further in Chapter 13.
Autoimmune Lymphoproliferative Syndrome.
Autoimmune lymphoproliferative syndrome (ALPS) is an autosomal dominant disease characterized by massive proliferation of lymphoid tissue with early lymphoma development. This genetic defect results in systemic autoimmune phenomenon (hence its name) and increased susceptibility to chronic viral infections only. Patients have an increased number of double-negative (CD4−CD8−) T cells but may eventually develop B-cell lymphomas. Most ALPS patients have a mutation in the gene coding for the Fas protein (CD95) (see Figure 18.3). Signaling through this protein normally activates apoptosis, or programmed cell death (see Chapters 11 and 13). Without activation of apoptosis, cells that should have died, such as autoimmune cells, continue to live, and immune responses that should have been turned off persevere. The unchecked proliferation of B cells then provides fertile ground for transforming mutations that lead to B-cell lymphomas. Most ALPS patients have one normal and one mutated Fas molecule, suggesting that the mutated Fas molecule interferes with the function of the normal molecule when they are cross-linked. Some ALPS patients have defects in other components of the apoptosis pathway, such as Fas ligand or caspase 10.
Two mouse strains—lpr and gld—have phenotypes similar to those of ALPS patients. The lpr mice have a mutation in their Fas gene and gld mice have a mutation in their Fas ligand gene. For many years, lpr mice were studied as a model for autoimmune disease, specifically systemic lupus erythematosus (SLE), before their Fas gene defect was discovered.
B-Cell–Associated or Immunoglobulin-Associated Immunodeficiency Disorders
B-cell-associated or immunoglobulin-associated immune diseases range from defective B-cell development with complete absence of all Ig classes to deficiencies in a single class or subclass of Ig. Patients suffer from recurrent or chronic infections that may start in infancy after maternal antibodies diminish (Bruton’s or X-linked agammaglobulinemia) or in young adulthood. Evaluation includes analysis of B-cell number and function and immunoelectrophoretic and quantitative determinations of Ig class and subclass.
X-Linked Agammaglobulinemia.
First described in 1952 by Bruton, X-linked agammaglobulinemia (XLA) is also called Bruton’s agammaglobulinemia. The disorder, which is relatively rare (1 in 100,000 individuals), is first noticed at approximately 6 months of age, when the infant has lost the maternally derived IgG that passed through the placenta. At that age, the infant presents with serious and repeated bacterial infections as a result of depression or virtual absence of all Ig classes.
The major defect lies in the inability of pre-B cells, which are present at normal levels in the bone marrow, to develop into mature B cells. The BTK gene, which is mutated in XLA, normally codes for a tyrosine kinase enzyme (Btk) residing in the cytosol. Btk is essential for signal transduction from the pre-B cell receptor (pre-BCR) on developing B cells. Without this signal, the cell develops no further (see Figure 18.1, defect 4). Only the nonmutated X chromosome is active in all mature B cells from female carriers of the mutant gene. XLA is therefore one of several inherited immunodeficiency diseases in which a mutation in a cytoplasmic tyrosine kinase is responsible for the disorder (mutations in JAK3 in SCID and ZAP-70 in T-cell deficiency are other examples).
Analyses of the blood, bone marrow, spleen, and lymph nodes of XLA patients reveal near absence of mature B cells and plasma cells, explaining the depressed Ig levels. Characteristically, affected children have markedly underdeveloped tonsils. The limited numbers of B cells appear normal in their ability to become plasma cells. Infants with XLA have recurrent bacterial otitis media, bronchitis, septicemia, pneumonia, arthritis, meningitis, and dermatitis. The most common microorganisms found to cause infections in these patients are Haemophilus influenzae and Streptococcus pneumoniae. Frequently, patients suffer from malabsorption due to infestation of the gastrointestinal tract with Giardia lamblia. Unexpectedly, XLA patients are also susceptible to infections by viruses that enter through the gastrointestinal tract, such as echovirus and polio. The infections do not respond well to antibiotics alone; treatment consists of periodic injections of intravenous γ-globulin (IVGG) containing large amounts of IgG (discussed further in Chapter 21). Although such passive immunization has maintained some patients for 20–30 years, the prognosis is guarded, as chronic lung disease due to repeated infections often supervenes.
Two other rare mutations, μ heavy-chain deficiency and λ5 chain deficiency, result in an identical clinical picture as a result of the same underlying pathophysiology. The λ5 chain is part of the surrogate light chain on pro-B cells and, along with the μ heavy chain and a third chain, forms the pre-BCR. Without a functional pre-BCR to receive signals, continued maturation of pro- to pre- B cells is halted and the cells die. Both of these mutations have autosomal recessive patterns of inheritance.
Transient Hypogammaglobulinemia.
When infants are approximately 5–6 months of age, passively transferred maternal IgG disappears, and IgG production by the infant begins to rise. Premature infants may have transient IgG deficiency if they are not yet able to synthesize Ig. Occasionally, a full-term infant may also fail to produce appropriate amounts of IgG, even when levels of IgM and IgA are normal. The cause appears to be a deficiency in number and function of helper T cells. Transient hypogammaglobulinemia may persist for a few months to as long as 2 years. It is not sex-linked and can be distinguished from the non-X-linked inherited agammaglobulinemias by the presence of normal numbers of B cells in the blood. Although treatment is usually not necessary, affected infants need to be identified, as they should not be given immunizations during this period.
Common Variable Immunodeficiency Disease.
Patients with common variable immunodeficiency disease (CVID) have markedly decreased serum IgG and IgA levels, with normal or low IgM and normal or low peripheral B-cell numbers. The cause of the disease, which affects both males and females, is not entirely clear and is probably not uniform. Onset may occur at any age, with peaks at 1–5 years old and 15–20 years old. Affected individuals suffer from recurrent respiratory and gastrointestinal infections with pyogenic bacteria and, paradoxically, from autoantibody-associated autoimmune diseases such as hemolytic anemia, autoimmune thrombocytopenia, and SLE. Many also have disorders of cell-mediated immunity. Long term, these patients have a high incidence of cancers, particularly lymphomas and gastric cancers.
CVID is characterized by a failure in the maturation of B cells into antibody-secreting cells (see Figure 18.1, defect 5). This defect may be due to an inability of the B cells to proliferate in response to antigen, normal proliferation of B cells without secretion of IgM, secretion of IgM without class switching to IgG or IgA (due to an intrinsic B-cell or T-cell abnormality), or failure of glycosylation of IgG heavy chains. In most cases, the disorder appears to be the result of diminished synthesis and secretion of Ig. The disease is familial or sporadic, with unknown environmental influences triggering onset. In the peripheral blood, patients will have normal or near-normal numbers of B cells but usually a marked reduction of memory B cells (CD27+/IgD− B cells). Memory B cells are those that have been exposed to antigen in the germinal center, where they have hypermutated and isotype switched their immunoglobulin genes and are now ready to respond rapidly with high-affinity antibody on reexposure to antigen.
Treatment depends on severity. For severe disease with many recurrent or chronic infections, IVGG therapy is indicated. Treated patients can have a normal life span. Women with CVID have normal pregnancies but, of course, do not transfer maternal IgG to the fetus.
Selective Immunoglobulin Deficiencies.
Several syndromes are associated with selective deficiency of a single class or subclass of Ig. Some are accompanied by compensatory elevated levels of other isotypes, such as the increased IgM levels in cases of IgG or IgA deficiency.
IgA deficiency is the most common immunodeficiency disorder in the Western world, with an incidence of approximately 1 in 800 individuals (see Figure 18.1, defect 6). The cause is unknown but appears to be associated with decreased release of IgA by B lymphocytes. IgA deficiency can also occur transiently as an adverse reaction to drugs. Patients may suffer from recurrent sinopulmonary viral or bacterial infections and/or celiac disease (defective absorption in the bowel); alternatively, they may be entirely asymptomatic. Isolated IgA deficiency may also precede full-blown expression of CVID; the two disorders are often found in the same families.
Treatment of symptomatic patients with IgA deficiency consists of broad-spectrum antibiotics. Therapy with immune serum globulin is not useful because commercial preparations contain only low levels of IgA, and injected IgA does not reach the areas of the secretory immune system where IgA is normally the protective antibody. Furthermore, patients may mount antibody responses (usually IgG or IgE) to the IgA in the transferred immune serum, causing hypersensitivity reactions. The prognosis for selective IgA deficiency is generally good, with many patients surviving normally.
Selective deficiencies in other Ig isotypes include IgM deficiency, a rare disorder in which patients suffer from recurrent and severe infections with polysaccharide-encapsulated organisms such as pneumococci and H. influenzae. Selective deficiencies in subclasses of IgG have been described but are even rarer.
Disorders of T–B Interactions
There are at least two diseases in which the T- and B-cell lineages appear to mature normally but demonstrate abnormal interactions between them. Although both disorders described below are usually due to underlying T-cell abnormalities, the predominant clinical symptoms are in the B-cell or humoral immune response.
Hyper-IgM Syndrome.
Patients with hyper-IgM syndrome (HIGM) present with recurrent respiratory infections at 1 to 2 years of age; very low serum IgG, IgA, and IgE; and normal to elevated IgM (see Figure 18.1, defect 8). In the most common form of the disease, the B cells are normal in number, functional in vitro, and will switch isotypes when appropriately stimulated. The T cells are also normal in number, subset distribution, and proliferative responses to mitogens. This form, X-HIGM, caused by a mutation in the CD40L gene on the X chromosome, results in the absence of CD40 ligand (CD154) on TH cells (see Figure 18.3). CD40L binds to CD40 expressed on B cells (see Chapter 11). This interaction is necessary for B-cell proliferation, class switching, and even germinal center formation. CD40–CD40L interaction also plays a role in macrophage–TH1 cell interaction; this aspect may explain the propensity of these patients to develop opportunistic infections, particularly pneumocystis pneumonia (PCP), and their poorer prognosis compared to XLA patients. For the same reason, delayed-type hypersensitivity (DTH) reactions are absent.

Full access? Get Clinical Tree


