INTRODUCTION
SUMMARY
Primary immune deficiency diseases (PIDDs) are characterized by increased susceptibility to infections, often associated with autoimmunity and inflammation and an increased risk of malignancies because of impaired immune homeostasis and surveillance. Depending on the nature of the immune defect, the clinical presentation of PIDD may vary and may include recurrence of upper and lower respiratory tract infections, invasive bacterial infections, purulent lymphadenitis, skin or deep abscesses, infections sustained by poorly virulent or opportunistic pathogens (Pneumocystis jirovecii, cytomegalovirus, environmental mycobacteria, Cryptosporidium, Giardia lamblia), persistent or recurrent candidiasis, narrow susceptibility to a selective type of pathogens, autoimmunity, increased susceptibility to malignancies, and may be associated with typical signs of specific immunodeficiency syndromes.
With the exception of immunoglobulin (Ig) A deficiency and DiGeorge syndrome, PIDDs are generally rare, with a prevalence of approximately 1 in 10,000 to 1 in 50,000 of the general population. However, prompt recognition of PIDD is of importance, because diagnostic delay is associated with increased risk of death and of irreversible complications. Most forms of PIDD follow mendelian inheritance; however, some, like common variable immunodeficiency (CVID), have a multifactorial origin. In most cases, PIDDs present in childhood, but late presentations may occur or even predominate in some forms, such as CVID.
The diagnostic approach to PIDD is based on a detailed family and clinical history, physical examination and appropriate laboratory tests. Lymphopenia is characteristic of severe combined immune deficiency. Abnormalities affecting neutrophils can be observed in patients with disorders of neutrophil production (e.g., congenital neutropenia, leukocyte adhesion deficiency; Chap. 65) or function (e.g., chronic granulomatous disease; Chap. 66), respectively. Evaluation of serum immunoglobulin levels and of antibody responses to immunization antigens is of value for patients with a history of recurrent infections. The clinical presentation and the results of these screening evaluations may prompt additional laboratory testing. For instance, patients with a profound hypogammaglobulinemia and a history of recurrent infections should be tested for the presence of circulating B lymphocytes (CD19+ or CD20+ cells), which are absent or markedly reduced in X-linked agammaglobulinemia. On the other hand, early presentation with severe and/or opportunistic infections, especially if associated with lymphopenia, should prompt enumeration of lymphocyte subsets. A severe reduction of circulating CD3+ T cells is typically observed in severe combined immune deficiency, and may be associated with defects of B and/or natural killer cells. Deep bacterial infections, or infections sustained by Aspergillus, require evaluation of neutrophil count and function, to identify patients with congenital neutropenia and chronic granulomatous disease, respectively. Invasive recurrent infections sustained by Neisseria species are an indication for assessing complement levels and function. The complement component deficiencies may also lead to systemic lupus erythematosus-like features or other autoimmune disorders. Laboratory results should be compared to age-matched control values, as white blood cell counts, lymphocyte subsets, complement components, immunoglobulin levels, and antibody production (especially to polysaccharide antigens) undergo significant changes and progressive maturation in the first years of life. It is important to rule out secondary forms of immunodeficiency, such as human immunodeficiency virus infection, protein loss, and immunodeficiency secondary to use of immunosuppressive drugs, as well as anatomical and/or functional problems (e.g., asplenia) that may lead to increased susceptibility to infections.
Recognition of PIDDs is essential to start optimal therapies at an early age. These include immunoglobulin substitution for patients with antibody deficiency; allogeneic hematopoietic stem cell transplantation for patients with severe combined immune deficiency; and in some cases, gene therapy or enzyme replacement therapy may be considered. Antimicrobial prophylaxis and aggressive treatment of infections is necessary in most cases of PIDD. Some patients with significant immune dysregulation may benefit from immunosuppressive therapy.
This chapter focuses on defects that primarily affect T and B lymphocytes, the complement system, and innate immunity. It discusses specific immunodeficiency syndromes, reviews etiology and pathogenesis, clinical and laboratory features, treatment, and prognosis. Chapters 65 and 66 discuss in detail disorders of neutrophil number and function.
Acronyms and Abbreviations
AD, autosomal dominant; ADA, adenosine deaminase; AD-HIES, autosomal dominant hyperimmunoglobulin E syndrome; aHSCs, autologous hematopoietic stem cells; AID, activation-induced cytosine deaminase; AIRE, autoimmune regulator; ALPS, autoimmune lymphoproliferative syndrome; APECED, autoimmune polyendocrinopathy, candidiasis, and ectodermal dystrophy; APS, autoimmune polyglandular syndrome; AR-HIES, autosomal recessive hyperimmunoglobulin syndrome; AT, ataxia-telangiectasia; ATLD, ataxia-telangiectasia–like disorder; ATM, ataxia-telangiectasia mutated; BCG, bacillus Calmette-Guérin; BLM, the causative gene of Bloom syndrome; BS, Bloom syndrome; BTK, Bruton tyrosine kinase; C, complement; CARD, caspase recruitment domain-containing protein; CD40L, CD40 ligand; CHARGE, coloboma of the eye, heart defects, atresia of the nasal choanae, retardation of growth and/or development, genital and/or urinary abnormalities, and ear abnormalities and deafness; CID, combined immune deficiency; CMC, chronic mucocutaneous candidiasis; CMV, cytomegalovirus; CSR, class switch recombination; CTL, cytotoxic T lymphocyte; CTLA-4, cytotoxic T-lymphocyte antigen-4; CTPS1, cytidine 5-triphosphate synthase 1; CVID, common variable immunodeficiency; D, diversity; DC, dendritic cell; DGS, DiGeorge syndrome; DOCK8, dedicator of cytokinesis 8; EBV, Epstein-Barr virus; FHL, familial hemophagocytic lymphohistiocytosis; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; GS2, Griscelli syndrome type 2; HIES, hyperimmunoglobulin E syndrome; HLA, human leukocyte antigen; HLH, hemophagocytic lymphohistiocytosis; HPV, human papillomavirus; HSCT, hematopoietic stem cell transplantation; HSE, herpes simplex virus encephalitis; HSV, herpes simplex virus; IBD, inflammatory bowel disease; ICF, immunodeficiency with centromere instability and facial anomalies; IFN, interferon; Ig, immunoglobulin; IGF-1, insulin-like growth factor 1; IGHM, immunoglobulin heavy constant mu; IGLL1, immunoglobulin lambda-like polypeptide 1; IKK, IκB kinase; IL, interleukin; IL-7R, IL-7 receptor; iNKT, invariant natural killer T cell; IPEX, immune dysregulation, polyendocrinopathy, enteropathy, X-linked; IRAK, IL-1 receptor-associated kinase; IRF8, interferon-regulated factor 8; ISG15, interferon-stimulated gene 15; ITCH, itchy E3 ubiquitin protein ligase; ITK, interleukin-2–inducible T-cell kinase; IVIG, intravenous immunoglobulin; J, joining; JAK3, Janus-associated tyrosine kinase 3; LCK, lymphocyte-specific protein tyrosine kinase; LGL, large granular lymphocytic leukemia; LIG4, DNA ligase IV; LRBA, lipopolysaccharide responsive beige-like anchor; LYST, lysosomal trafficking regulator; MAGT1, magnesium transporter 1; MHC, major histocompatibility complex; MMF, mycophenolate mofetil; MonoMAC, monocytopenia, B-cell and NK-cell lymphopenia associated with mycobacterial, fungal, and viral infections; MSMD, mendelian susceptibility to mycobacterial disease; MyD88, myeloid differentiation factor 88; NBS, Nijmegen breakage syndrome; NEMO, nuclear factor-κB essential modulator; NF, nuclear factor; NK, natural killer; NKT, natural killer T cell; ORAI1, calcium release-activated calcium channel protein 1; PI3K, phosphatidylinositol 3-kinase; PIDD, primary immune deficiency disease; PLDN, pallidin; PMS2, postmeiotic segregation increased 2 (Saccharomyces cerevisiae); PNP, purine nucleoside phosphorylase; RAG1/2, recombination activating gene 1/2; RMRP, ribonuclease mitochondrial RNA processing complex; SAP, signaling lymphocyte activation molecule (SLAM)-associated protein; SCID, severe combined immune deficiency; SHM, somatic hypermutation; SLAM, signaling lymphocyte activation molecule; SMARCAL1, switch/sucrose nonfermentable (SWI/SNF)-related matrix-associated actin-dependent regulator of chromatin subfamily A–like protein 1; SNP, single nucleotide polymorphism; STIM1, stromal interaction molecule 1; TAP-1/2, transport-associated protein 1/2; TCR, T-cell receptor; TEMRA, T memory; TLR, toll-like receptor; TNF, tumor necrosis factor; TRAF, TRIF-related adaptor molecule; TREC, T-cell–receptor excision circle; TRIF, toll-interleukin 1 receptor domain-containing adaptor-inducing IFN-β; TYK2, tyrosine kinase 2; UNG, uracil N-glycosylase; V, variable; VODI, venoocclusive disease with immunodeficiency; WAS, Wiskott-Aldrich syndrome; WASp, Wiskott-Aldrich syndrome protein; WHIM, warts, hypogammaglobulinemia, infections, myelokathexis; WIP, WASp-interacting protein; WRN, Werner syndrome, RecQ helicase-like; XHIGM, X-linked hyperimmunoglobulin M; XLA, X-linked agammaglobulinemia; XLP1 and XLP2, X-linked lymphoproliferative syndrome types 1 and 2; XLT, X-linked thrombocytopenia; ZAP-70, zeta-associated protein of 70 kDa.
PREDOMINANT ANTIBODY DEFICIENCIES
X-linked agammaglobulinemia (XLA) is the prototypic antibody deficiency characterized by profound hypogammaglobulinemia caused by a maturation defect in B-cell development.1,2 XLA, originally described in 1953, is one of the first primary immunodeficiencies in which the underlying defect, a mutation of Bruton tyrosine kinase (BTK) was identified. Autosomal recessive agammaglobulinemia, a variant form of agammaglobulinemia, has been reported in patients with a clinical phenotype resembling XLA including very low B-cell numbers and severe bacterial infections but normal BTK.2 Several responsible gene mutations have been identified, including those involving the B-cell receptor complex μ heavy chain (immunoglobulin heavy constant mu [IGHM]), the surrogate light chain component λ5 (immunoglobulin lambda-like polypeptide 1 [IGLL1]), the signal transducer complex of the pre–B-cell receptors immunoglobulin (Ig) α (CD79a), Igβ (CD79b), and mutations in the B-cell adaptor molecule BLINK, the p85α subunit of phosphatidylinositol 3-kinase (PI3K)3 and a dominant negative E47 mutation causing autosomal dominant agammaglobulinemia.4
Because IgG is actively transported across the placenta, infants born with XLA have normal levels of IgG at birth and are frequently asymptomatic for the first few months of life. Following metabolism of the maternal antibodies, affected boys begin to develop recurrent infections usually between 4 and 12 months of age. In a review of 96 XLA patients, 20 percent experienced initial clinical symptoms after their first birthday and approximately 10 percent after 18 months of age.5 In an Italian study of 73 patients with mutation-verified XLA, the mean age of onset of symptoms was 2 years.6 The presenting symptoms vary greatly and may be mild or severe (Table 80–1). Otitis media and chronic sinusitis, pneumonia, pyoderma, and diarrhea are frequent clinical presentations. Serious complications include septicemia, meningitis, septic arthritis, and osteomyelitis. In young children with XLA, acute infections are often associated with neutropenia. Pyogenic bacteria, such as Haemophilus influenzae, Streptococcus pneumoniae, and Staphylococcus aureus are the most common pathogens observed in XLA. Opportunistic infections, such as Pneumocystis jirovecii, are rarely observed. Infections with Ureaplasma urealyticum have been reported in XLA patients with mycoplasma arthritis.7 Although resistance to viral infections is generally intact, XLA patients are unusually susceptible to enteroviruses such as echovirus, coxsackievirus, and poliovirus. Poliomyelitis after live-attenuated (Sabin) poliovirus vaccine, especially if given at a time when maternal antibodies had disappeared, is associated with high morbidity and mortality.5 Before the introduction of intravenous immunoglobulin (IVIG), XLA patients frequently developed chronic, disseminated echovirus and coxsackievirus infections presenting as meningoencephalitis, dermatomyositis/fasciitis, and hepatitis.8 Gastroenteritis caused by Giardia lamblia, Campylobacter species, or rotavirus is not uncommon and may be associated with malabsorption. Chronic intestinal inflammation resembling Crohn disease may develop in children and adults with XLA. Interestingly, an increased incidence of rectosigmoid cancer with high mortality has been reported.9 Pyoderma gangrenosum-like ulcers of the lower extremities have been observed to be caused by Helicobacter species.10
| Neutrophil Numerical or Functional Defects (See Chaps. 65, 66) | Antibody Deficiencies | Combined Immune Deficiencies | Complement Deficiencies |
|---|---|---|---|
| Severe bacterial and fungal infections | Recurrent bacterial infections after 4 to 6 months of age | Early onset respiratory and gut infections (bacterial, viral, fungal) | Recurrent or severe infections sustained by encapsulated pathogens |
| Skin or deep bacterial and fungal abscesses | Intestinal Giardia lamblia infection | Opportunistic infections | Recurrent Neisseria meningitidis infections |
| Infections sustained by unusual bacteria and fungi | Enteroviral meningoencephalitis | Persistent candidiasis Erythroderma | Autoimmune manifestations (systemic lupus erythematosus-like) |
| Colitis | Growth failure | Atypical hemolyticuremic syndrome | |
| Recurrent angioedema (C1-INH deficiency) |
Most patients have markedly reduced levels of all classes of immunoglobulins; circulating B cells are less than 1 percent of total lymphocytes and tonsils are absent. Because of the maturation arrest at the pre–B-cell stage, very few B cells undergo differentiation into plasma cells. As a result, lymph nodes, lymphoid follicles, germinal centers, and intestinal mucosal biopsies lack plasma cells. As expected, specific antibodies to microorganisms or vaccines are markedly reduced or undetectable (Table 80–2).
| Humoral Immunity | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lymphocytes* | Serum Immunoglobulins (Ig) | |||||||||
| B | T | NK | Cellular Immunity | M | G | A | E | Antibody Responses | Common Infections | |
| Predominantly antibody deficiencies | ||||||||||
X-linked agammaglobulinemia BTK deficiency | − | + | + | + | ↓ | ↓ | ↓ | ↓ | − | Bacteria, Giardia lamblia |
| Autosomal recessive agammaglobulinemia | ||||||||||
| λ5, Igα, Igβ, BLNK, p85α, E47 deficiency | − | + | + | + | ↓ | ↓ | ↓ | ↓ | − | Bacteria |
| Transient hypogammaglobulinemia of infancy | + | + | + | + | N/↓ | N/↓ | N/↓ | N/↓ | +/− | Bacteria |
| Selective IgA deficiency | + | + | + | + | N | N | ↓ | N | +/− | Bacteria, G. lamblia |
| Common variable immune deficiency (CVID) | + | + | + | + | N/↓ | ↓ | ↓ | ↓ | − | Bacteria, G. lamblia |
| Hyper-IgM syndromes | ||||||||||
| CD40 ligand deficiency (X-linked) | + | + | + | +/− | N/↑ | ↓ | ↓ | ↓ | +/− | Bacteria, viruses, fungi |
| CD40 deficiency | + | + | + | + | N/↑ | ↓ | ↓ | ↓ | +/− | Bacteria, viruses, fungi |
| Activation-induced cytidine deaminase deficiency (AID) | + | + | + | + | N/↑ | ↓ | ↓ | ↓ | +/− | Bacteria |
| Uracil-DNA glycosylase deficiency (UNG) | + | + | + | + | N/↑ | ↓ | ↓ | ↓ | +/− | Bacteria |
| X-linked NF-κB Essential Modulator (NEMO) deficiency, due to mutations in IKBKG | + | + | + | + | N/↑ | ↓ | ↓ | ↓ | +/− | Bacteria, viruses, fungi |
| Severe combined immunodeficiencies (SCID) | ||||||||||
| Interleukin receptor γ-chain deficiency (X-linked SCID) | + | − | − | − | N | ↓ | ↓ | ↓ | − | Bacteria, viruses, fungi |
| Janus-associated kinase 3 (JAK3) deficiency | + | − | − | − | N | ↓ | ↓ | ↓ | − | Bacteria, viruses, fungi |
| Interleukin-7 receptor α-chain deficiency | + | − | + | − | N | ↓ | ↓ | ↓ | − | Bacteria, viruses, fungi |
| ZAP-70 tyrosine kinase deficiency | + | +/− | + | − | N | N/↓ | N/↓ | N/↓ | +/− | Bacteria, viruses, fungi |
| Adenosine deaminase (ADA) deficiency | − | − | − | − | ↓ | ↓ | ↓ | ↓ | − | Bacteria, viruses, fungi |
| Purine nucleotide phosphorylase (PNP) deficiency | + | − | + | − | N | ↓ | ↓ | ↓ | +/− | Bacteria, viruses, fungi |
| Recombinase activating genes (RAG 1/2) deficiency | − | − | + | − | ↓ | ↓ | ↓ | ↓ | − | Bacteria, viruses, fungi |
| Artemis deficiency | − | − | + | − | ↓ | ↓ | ↓ | ↓ | − | Bacteria, viruses, fungi |
| Reticular dysgenesis (AK2 deficiency) | − | − | − | − | ↓ | ↓ | ↓ | ↓ | − | Bacteria, viruses, fungi |
| Primary T-cell deficiencies | ||||||||||
| Congenital thymic aplasia (DiGeorge syndrome) | + | − | + | +/− | N | N | N | N | +/− | Bacteria, viruses, fungi |
| MHC class II deficiency | + | +/− | + | + | N/↓ | ↓ | N/↓ | ↓ | +/− | Bacteria, viruses, fungi |
| TAP-1, TAP-2 deficiency (MHC class I deficiency) | + | +/− | +/− | +/− | N | N | N | N | +/− | Bacteria, viruses, fungi |
| Other well-defined immunodeficiency syndromes | ||||||||||
| Ataxia-telangiectasia | + | + | + | +/− | N/↑ | N/↓ | N/↓ | ↓ | +/− | Bacteria |
| Wiskott-Aldrich syndrome | + | +/− | + | +/− | ↓ | N | ↑ | ↑ | +/− | Bacteria |
| Hyper IgE Syndromes | ||||||||||
| STAT3 deficiency (AD) | +/− | + | + | +/− | N | N | N | ↑↑ | +/− | Staph, Candida |
| DOCK8 deficiency (AR) | +/− | +/− | +/− | +/− | ↓ | N | N | ↑↑ | +/− | Candida, viruses, fungi |
| GATA 2 deficiency (AD) | − | + | − | +/− | N | N | N | N | +/− | Atypical mycobacteria, viruses, fungi |
| IPEX, IPEX-like | + | (lack of Tregs) | + | + | N | N | ↑ | ↑ | + | Autoimmunity, Staph, Candida, CMV |
BTK, a cytoplasmic protein tyrosine kinase known to interact with other cytoplasmic proteins, plays an important role in the pre–B-cell expansion and the survival of mature B cells by facilitating signaling through the B-cell antigen receptor. BTK is present in all hematopoietic cells except T cells, natural killer (NK) cells, and plasma cells. The presence of BTK in normal monocytes and platelets allows assessment of BTK in most XLA patients with low or absent BTK levels using flow cytometry, and to identify carrier females.11 Sequence analysis of the BTK gene confirms the diagnosis of XLA and allows prenatal diagnosis. Autosomal recessive (and dominant) forms of agammaglobulinemia are rare and require sequence analysis of the genes listed above.
Intravenous or subcutaneous IgG infusions at a dose of 400 to 600 mg/kg every 3 to 4 weeks are highly effective in preventing chronic infections in agammaglobulinemic patients. Prophylactic antibiotics are indicated in those with chronic lung disease. Adequate IVIG replacement has markedly reduced the incidence of enteroviral infections, but other complications, such as Crohn-like disease, are difficult to prevent, and progressive neurodegeneration has been observed in a small number of XLA patients without identification of an infectious agent.12
Hyper-IgM syndromes are characterized by recurrent infections associated with low serum levels of IgG, IgA, and IgE, but normal or increased levels of IgM (see Table 80–2). They are the direct result of mutations affecting genes involved in B-cell activation, class switch recombination (CSR), and somatic hypermutation (SHM). Mutations in the genes encoding CD40 ligand (CD40L) or CD40 interfere with the triggering of events that lead to CSR and SHM. Mutations in the B-cell intrinsic enzymes, activation-induced cytosine deaminase (AID) and uracil N-glycosylase (UNG), directly affect CSR and SHM. Mutations in the NEMO gene (nuclear factor [NF]-κB essential modulator), a protein crucial for NF-κB activation, cause clinical features of anhydrotic ectodermal dysplasia with associated immune deficiency in males and incontinentia pigmenti in females.13 A novel B-cell–intrinsic CSR deficiency characterized by susceptibility to malignancies was found to be associated with mutations in the gene encoding the postmeiotic segregation increased 2 (Saccharomyces cerevisiae) (PMS2) component of the mismatch repair machinery.14 A subset of patients with ataxia telangiectasia present with elevated serum IgM and CSR deficiency.15
Clinical Features In addition to recurrent bacterial infections, affected infants with X-linked hyperimmunoglobulin M (XHIGM) often present with interstitial pneumonia caused by P. jirovecii approximately 50 percent of affected males will develop neutropenia.16 Patients with XHIGM are at high risk of developing chronic Cryptosporidium infections complicated by ascending cholangiolitis and chronic liver disease. Progressive neurodegeneration in XHIGM patients similar to those with XLA has been reported.12 Abortive germinal center formation and severe depletion of follicular dendritic cells of lymph nodes occurs. Affected patients are at risk to develop neoplasms, most often lymphomas, but also tumors of the biliary and gastrointestinal tract,17 which are rarely observed in other primary immunodeficiencies.
Laboratory Features Circulating lymphocyte subsets are present in normal numbers but B cells are predominantly naïve and few are of the switched memory B-cell subtype (IgD–, IgM– CD27+).18 Lymphocyte proliferation in response to mitogens is normal, but responses to specific antigens are often reduced.19 XHIGM is caused by mutations in CD40L, a surface protein expressed by activated CD4+ lymphocytes. CD40L interacts with the CD40 membrane protein constitutively expressed by B cells, macrophages, and dendritic cells (DCs). The interaction of CD40L/CD40 sets in motion a signaling event that results in the expression of AID and UNG, and induces CSR and SHM. Mutations in CD40L are distributed throughout the gene and may result in nonfunctional or absent protein.16 Several patients with mild cases of XHIGM not treated with IVIG have developed chronic pure red cell aplasia as a result of persistent parvovirus B19 infection.20
Treatment Prophylactic treatment with trimethoprim-sulfamethoxazole is indicated during infancy and childhood to prevent P. jirovecii pneumonia. Intravenous or subcutaneous immunoglobulin at doses similar to patients with XLA is used to prevent chronic infections, including parvovirus B19. Exposure to Cryptosporidium should be prevented by avoiding the use of potentially contaminated water. Because of the high incidence of serious complications and the unfavorable long-term outcome,21 allogeneic stem cell transplantation should be considered if an optimal donor can be identified. Severe and persistent neutropenia may require treatment with granulocyte colony-stimulating factor (G-CSF), at least on a temporary basis.
Autosomal recessive hyperimmunoglobulin M caused by mutations in CD40 have been reported, mostly in consanguineous families.16,22 Affected members have similar clinical and laboratory findings as those with CD40L mutations. Treatment and prognosis of CD40 deficiency is similar to XHIGM.
Definition AID is expressed only in B cells undergoing CSR or SHM and is thought to affect DNA editing.23 Because of milder symptoms, the diagnosis of AID deficiency is often established later in life.
Clinical Features AID-deficient patients present with recurrent bacterial infections, mostly affecting the upper and lower respiratory tract. In contrast to patients with XHIGM, AID-deficient individuals have an excellent long-term prognosis, especially if given IVIG prophylaxis. Most affected individuals present with striking lymphoid hyperplasia involving tonsils and lymph nodes as a result of marked follicular hyperplasia. The number of circulating T- and B-cell subsets are normal, including normal proportion of memory B cells; however, all CD27+ memory B cells fail to isotype switch and only express IgM and IgD. Mutations of AID affect the entire gene and include missense, nonsense mutations, and small deletions.
UNG is expressed in proliferating cells, including B cells undergoing CSR. Following AID-induced deamination of cytosine into uracil residues on single-stranded DNA, UNG deglycosylates and removes uracil residues, thus leading to a single-stranded DNA break. The repair of the DNA nick leads to successful CSR and SHM. Because AID and UNG are functionally closely linked, lack of UNG results in a clinical phenotype similar to AID deficiency. The three UNG deficient patients reported to date have a history of frequent bacterial infections, lymphadenopathy, and an excellent response to IVIG therapy.23
X-Linked Anhydrotic Ectodermal Dysplasia with Immunodeficiency Caused by Mutations in Nuclear Factor-κB Essential Modulator
Definition Anhydrotic (or hypohidrotic) ectodermal dysplasia is a rare syndrome with partial or complete absence of sweat glands, sparse hair growth, and abnormal dentition. A subset of these patients has an X-linked mode of inheritance and immunodeficiency characterized by low-serum IgG levels, variably elevated IgM levels, and decreased antibody responses. This syndrome results from mutations in the IKBKG gene encoding NEMO, a key subunit of I-κB kinase that regulates NF-κB dimerization and nuclear transfer.24 Most affected boys have a hypomorphic NEMO mutation that allows some function, and present with bacterial (S. pneumoniae, S. aureus) and often atypical mycobacterial infections. Loss-of-function mutations cause the X-linked dominant condition of incontinentia pigmenti in females and are embryonically lethal in males. A similar phenotype with autosomal dominant inheritance is caused by gain-of-function mutations in IKBA.25
Clinical Features A review of 72 individuals with NEMO mutations has demonstrated a wide spectrum of clinical phenotypes.26 Thirty-two different mutations of NEMO were identified, with 70 percent being associated with ectodermal dysplasia, 86 percent with serious pyogenic infections, 39 percent with mycobacterial infections, 19 percent with serious viral infections, and 21 percent with inflammatory bowel disease. One-third of this cohort of NEMO patients died prematurely (mean age: 6.4 years).
Treatment Treatment with IVIG is useful but does not prevent the occurrence of serious complications. Symptomatic treatment depends on those complications.
Common variable immunodeficiency (CVID) is a clinically and molecularly heterogeneous disorder, presenting at any age, but most often during adulthood. CVID is characterized by recurrent bacterial infections, hypogammaglobulinemia, and impaired antibody responses. Together with selective IgA deficiency, CVID is the most common primary immune deficiency, with an incidence of 1 in 10,000 individuals. Familial inheritance is observed in approximately 20 percent of cases and CVID and IgA deficiency may be present in the same families. In rare instances, patients with selective IgA deficiency may progress to CVID. Attempts have been made to associate CVID and IgA deficiency with genes located within the major histocompatibility complex (MHC) region on chromosome 6; however, no specific genes within this region have been identified. A small proportion of patients with CVID have been molecularly defined as having mutations in several genes involved directly or indirectly with B-cell differentiation, including ICOS, TACI, BAFF-receptor, CD19, CD20, CD21, and CD81.27 The recent discovery of CVID-like phenotypes resulting from heterozygous mutations in NF-κB228,29 and gain-of-function mutations in PI3Kδ,30 and CVID with autosomal recessive inheritance as a result of mutations in protein kinase Cδ31 further support the idea that CVID is a heterogeneous primary immune deficiency disease (PIDD) with strong genetic roots. In addition, patients with mutations in BTK, CD40L, and SH2D1A have been mistakenly diagnosed as CVID.
The majority of CVID patients present with recurring sinopulmonary infections, most often bacterial pneumonia.27,32 If the diagnosis is delayed or if treatment is inadequate, bronchiectasis and chronic lung disease may develop. Gastrointestinal complaints are frequent and may be caused by chronic G. lamblia or Campylobacter infections, resembling chronic inflammatory bowel disease. Lymphoid hyperplasia of the small bowel is a frequent finding. Autoimmune disorders are common and may resemble rheumatoid arthritis, dermatomyositis, or scleroderma. In addition, CVID patients may develop autoimmune hemolytic anemia, autoimmune thrombocytopenia, autoimmune neutropenia, pernicious anemia, and chronic active hepatitis. Lymphadenopathy and splenomegaly are common, the result of follicular hyperplasia. Caseating granulomas of the lung, spleen, liver, skin, and other tissues may develop at any age, and a condition resembling sarcoidosis has been described. The cause of this devastating granuloma formation is unknown. A high incidence of lymphoma and gastrointestinal malignancies have been reported in older CVID patients,33 with a 438-fold increase in the risk of lymphomas in affected women during the fifth and sixth decades.34 Despite normal numbers of blood B lymphocytes and the presence of lymphoid cortical follicles, CVID patients have hypogammaglobulinemia that may be as profound as in XLA (see Table 80–2). Antibody responses to recall and to neoantigens are diminished and some CVID patients have decreased numbers of memory B cells, especially of switched memory B cells. A subset of CVID patients have a substantial T-cell deficiency characterized by decreased expression of CD40L by activated CD4+ T cells (without a mutation of CD40L) and by reversed CD4:CD8 ratio. Treatment with IVIG substitution and prophylactic antibiotics is beneficial but often insufficient to prevent serious complications. Allogeneic hematopoietic stem cell transplantation (HSCT) is generally not recommended, except in patients with lymphoid malignancies. There is a rare association between immune deficiency and thymoma, which is estimated to be present in 4 percent of patients with hypogammaglobulinemia.35
The incidence of selective IgA deficiency, defined as IgA less than 5 to 10 mg/dL, differs greatly between ethnic groups, being highest in Scandinavia (1 in 396 in a Finnish study)36 and lowest in Asian populations (1 in 14,000 in Japan).37 Because secretory IgA is considered to be most important in protecting mucus surfaces, it is surprising that most IgA-deficient patients remain healthy. Other defense systems, for example, noncirculatory IgM or neutrophils, may compensate for this deficiency. Symptomatic individuals are not only IgA deficient, but often have deficient antibody responses to specific antigens. IgA deficiency may be associated with IgG2 and IgG3 deficiency and poor responses to polysaccharide antigens.38 Selective IgA deficiency, if associated with symptoms, often leads to recurrent sinopulmonary infections and atopic symptoms including allergic conjunctivitis, rhinitis, and eczema. Food allergy may be more common in IgA-deficient patients and asthma associated with IgA deficiency appears to be more refractory to therapy. Gastrointestinal tract disorders include chronic giardiasis, malabsorption, celiac disease, primary biliary cirrhosis, pernicious anemia, and nodular lymphoid hyperplasia. A number of autoimmune diseases are associated with selective IgA deficiency, including rheumatoid arthritis, systemic lupus erythematous, thyroiditis, myasthenia gravis, and ulcerative colitis.
A significant proportion of IgA-deficient individuals has anti-IgA antibodies in their serum and may react to blood products containing IgA, including IVIG preparations with low IgA content. However, patients with selective IgA deficiency who make normal IgG antibody do not need IVIG therapy.
The fundamental defect in selective IgA deficiency is the failure of IgA-bearing B lymphocytes to mature into IgA-secreting plasma cells. There is no specific treatment that would correct this problem. Intermittent or continuous prophylactic antibiotics may be helpful in patients with recurrent respiratory tract infections, who develop chronic symptoms of lung disease. On the other hand, if IgA deficiency is associated with poor antibody responses to selected antigens, for example, to polysaccharides, an attempt with IVIG substitution should be made.
Lipopolysaccharide responsive beige-like anchor (LRBA) is a broadly expressed, cytosolic protein involved in endocytosis of ligand-activated receptors. LRBA deficiency is inherited as an autosomal recessive trait and is characterized by recurrent bacterial and viral infections, and prominent autoimmune manifestations, cytopenias, and inflammatory bowel disease, in particular.39,40 Hypothyroidism and myasthenia gravis have been also reported. Immunologic abnormalities include progressive hypogammaglobulinemia, impaired activation and decreased survival of T and B lymphocytes, reduced number of marginal zone-like and switched memory B cells, and defective autophagy.
SEVERE COMBINED IMMUNODEFICIENCIES
The first description of severe combined immunodeficiencies (SCIDs) dates back to 1950, when Glanzmann and Riniker described infants who died with overwhelming infections, intractable diarrhea, thrush, and profound lymphophenia.41 The SCID phenotype represents a heterogeneous group of genetic disorders that are characterized by a severe impairment of T-lymphocyte development and function (Fig. 80–1).42,43,44 Depending on whether the development of B and/or NK lymphocytes is also affected, SCID can be classified into four distinct immunologic phenotypes: (1) T–B+NK– SCID (the most common variant); (2) T–B+NK+ SCID; (3) T–B–NK+ SCID; or (4) T–B–NK– SCID (see Table 80–2). The term combined immune deficiency (CID) is used to define disorders with residual development and/or function of T lymphocytes. Unless treated by allogeneic HSCT or, in selected cases, by gene therapy or enzyme replacement therapy, SCID is inevitably fatal.
Figure 80–1.
Disruption of the normal T-cell development by mutations of genes known to cause a severe combined immunodeficiency disease phenotype. B, B lymphocyte; CLP, common lymphocyte progenitor; γc, common gamma; Gran-P, granulocyte progenitor; HSC, hematopoietic stem cell; JAK3, Janus-associated tyrosine kinase 3; MHC, major histocompatibility complex; Mono-P, monocyte progenitor; NK, natural kill lymphocyte; ORAI1, calcium release-activated calcium channel protein 1; T, T lymphocyte; TAP-1/2, transport-associated protein 1/2; ZAP70, zeta-associated protein of 70 kDa.
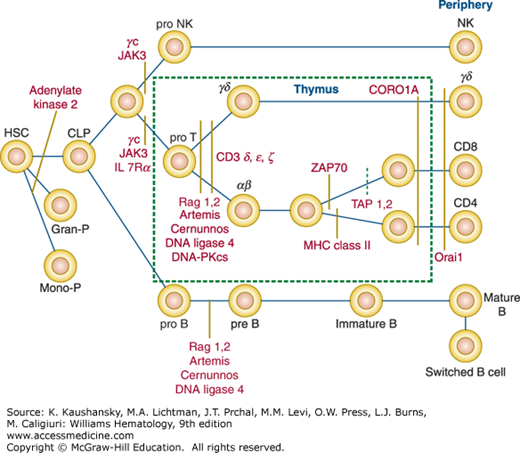
SCIDs are mendelian disorders, and their overall prevalence is estimated to be approximately 1 in 50,000 births. In Western countries, the most common form of SCID is inherited as an X-linked trait; however, a variety of autosomal recessive forms are also known. SCID can be grouped in different categories that illustrate the various pathogenetic mechanisms involved in T-cell development.
Adenosine Deaminase Deficiency Approximately 5 to 10 percent of infants with SCID have a deficiency of adenosine deaminase (ADA), the enzyme that converts adenosine and deoxyadenosine into inosine and deoxyinosine, respectively.45 In the absence of ADA, high intracellular levels of adenosine, deoxyadenosine, and their toxic phosphorylated metabolites cause apoptosis of lymphoid precursors, and hence result in the virtual absence of T lymphocytes, that is usually associated with marked reduction of B and NK lymphocytes (T–B–NK–SCID).46 ADA-SCIDis inherited as an autosomal recessive trait, and its clinical manifestations extend beyond the immune system, reflecting the fact that ADA is a general housekeeping enzyme.
Purine Nucleoside Phosphorylase Deficiency Purine nucleoside phosphorylase (PNP) is another enzyme of the purine salvage pathway. PNP catalyzes the phosphorylation of inosine, guanosine, and deoxyguanosine.45 In the absence of PNP, high intracellular levels of deoxyguanosine triphosphatase cause lymphoid and neuronal toxicity. Immature thymocytes are particularly susceptible to PNP deficiency. Accordingly, the immunologic phenotype of PNP deficiency is characterized by decreased T-cell counts, whereas B and NK lymphocytes are often unaffected.47 PNP deficiency accounts for 1 to 2 percent of all forms of SCID, and is inherited as an autosomal recessive trait.
Adenylate Kinase 2 Deficiency Another rare variant of autosomal recessive SCID, reticular dysgenesis, is characterized by extreme lymphopenia, absence of neutrophils, and sensorineural deafness.48 The disease is caused by mutations of adenylate kinase 2 that result in apoptosis of myeloid precursors of neutrophils, and of lymphoid progenitor cells.49,50
Thymic T-cell progenitors depend on interleukin (IL)-7 for cell proliferation. The IL-7 receptor (IL-7R) is composed of an α chain (encoded by the IL7R gene) and a common γ chain (γc), that is shared also by IL-2R, IL-4R, IL-9R, IL-15R, and IL-21R,51 and is encoded by the IL2RG gene, located on the X chromosome. Cytokine-mediated signaling through γc—containing receptors involves activation of Janus-associated tyrosine kinase 3 (JAK3).51 In humans, defects of IL-7–mediated signaling abrogate T-cell development, whereas impaired signaling through IL-15R affects development of NK cells.51 X-linked SCID, caused by IL2RG mutations,52 represents approximately 30 percent of all cases of SCID, and is characterized by lack of T and NK lymphocytes but normal development of B cells (T–B+NK– SCID). B-lymphocyte function, however, is severely compromised by both the lack of T-cell help and nonfunctional γc. JAK3 deficiency is inherited as an autosomal recessive trait, and its phenotype is identical to that of X-linked SCID (T–B+NK– SCID).53,54 In contrast, autosomal recessive IL-7R deficiency caused by mutation of the α chain is characterized by the selective lack of T cells (T–B+NK+ SCID).55
One of the distinctive features of developing thymocytes is the expression of the pre–T-cell receptor (TCR), that is composed of a pre-Tα chain, a TCRβ chain, and the CD3 γ, δ, ε, and ζ chains. Signaling through the pre-TCR permits rearrangement of the TCRα chain and expression of a mature TCRαβ. Alternatively, thymocytes may express the γδ chains of the TCR. Rearrangement of the TCR loci is accomplished by means of the V(D)J recombination, whereby the lymphoid specific recombination activating gene 1 (RAG1) and recombination activating gene 2 (RAG2) proteins mediate DNA cleavage at the variable (V), diversity (D), and joining (J) elements of the TCR loci. The DNA double-strand break of the coding ends is initially sealed as a hairpin, followed by nonhomologous endjoining via the nuclease Artemis (encoded by the DCLRE1C gene). Eventually, joining of coding (and signal) elements is mediated by a series of proteins, that include the Ku70/80 heterodimer, XRCC4, DNA ligase IV (LIG4), DNA-protein kinase catalytic subunit, and Cernunnos/XLF. Defects in V(D)J recombination affect both T- and B-cell development and hence cause T–B–NK+ SCID, because this process is also essential to mediate rearrangement of the immunoglobulin genes, a key step in B-cell development. RAG1 or RAG2 deficiencies account for 3 to 20 percent of all SCID cases in different series.42,56 Artemis (DCLRE1C),57 DNA-protein kinase catalytic subunit,58 LIG4,59,60 and Cernunnos/XLF61 deficiencies are less frequent and their cellular and clinical phenotypes extend beyond impaired T- and B-cell development, because enzymes that mediate DNA double-strand break repair are ubiquitously expressed, and their deficiency results in increased cellular radiosensitivity.57,58,59,60,61 The phenotype of LIG4 deficiency can be extremely variable, from T–B–NK+ SCID to mild or no immunodeficiency, whereas Cernunnos/XLF deficiency is characterized by significant T-cell lymphopenia and progressive decrease in the number of B cells.
Defects of the CD3 δ, ε, or ζ chains affect signaling through the pre-TCR and the TCR and hence cause autosomal recessive T–B+NK+ SCID.62,63,64 In contrast, CD3γ deficiency is associated with mild T-cell lymphopenia and a variable clinical phenotype.65,66
Mutations of the TCRα constant (TCRA) gene cause impaired differentiation of T cells expressing TCRαβ.67
Mutations of CD45, a pan-leukocyte tyrosine phosphatase that has been implicated in signaling through the TCR and the B-cell receptor, have been reported in few patients with T–B+NK+ SCID.68,69
Despite genetic heterogeneity, SCID is characterized by a consistent clinical phenotype. Interstitial pneumonia, often sustained by P. jirovecii, cytomegalovirus (CMV), adenovirus, parainfluenza 3 virus, respiratory syncytial virus, chronic diarrhea, failure to thrive, and persistent candidiasis are common features (see Table 80–1). Typically, infections develop in the first months of life. Skin manifestations (maculopapular rash, erythroderma, alopecia) are also common, especially in infants with maternal T-cell engraftment. Hypoplastic lymphoid tissue (tonsils, lymph nodes), and absence of a thymic shadow on chest radiography are characteristic.70
Because of the inability to control replication of live microorganisms, administration of live-attenuated vaccines often leads to severe, life-threatening complications in infants with SCID.71
T-cell engraftment derived from maternal cells that cross the placenta occurs in more than 50 percent of infants with SCID. Most often asymptomatic, it may cause skin rash or, less frequently, typical graft-versus-host disease with generalized rash, liver disease, profuse diarrhea, jaundice, and severe hematologic abnormalities (thrombocytopenia, anemia, leukopenia) that are indicative of marrow damage.72,73 Transfusion of un-irradiated blood products often leads to fatal graft-versus-host disease.
An absolute lymphocyte count less than 2000/μL should prompt immediate investigation for SCID, regardless of the severity of clinical symptoms.42 Typically, infants with SCID have markedly reduced or absent circulating T cells which are unable to proliferate in vitro in response to mitogens and specific antigens.74 However, T lymphocyte count may be preserved, at least in part, in SCID infants with maternal T-cell engraftment, with “leaky” variants of the disease,74 or with somatic reversions that allow for some autologous T-cell development.75 Finally, T-lymphocyte count can be normal or modestly reduced in patients with functional T-cell immunodeficiencies (see other combined immunodeficiencies; defective thymic development).
Maternal T-cell engraftment and “leaky” SCID with residual development of autologous T cells are characterized by the expression of the CD45R0 memory/activation antigen on the surface of circulating T lymphocytes (whereas most T cells in normal infants have a naïve CD45RA+ phenotype).
TCR excision circles, consisting of circularized signal joints, are a byproduct of V(D)J recombination and are exported to the blood by recent thymic emigrants. Levels of TCR excision circles in circulating lymphocytes are particularly high in newborns and infants, and progressively decline with age. Because TCR excision circles cannot be detected in infants with SCID, assessment of TCR excision circle levels by polymerase chain reaction has been successfully introduced for newborn screening for SCID.76
Although the number of circulating B lymphocytes can vary depending on the nature of the genetic defect, serum immunoglobulin levels are low in infants with SCID (see Table 80–2). Normal serum IgG levels early in life reflect transplacental passage of maternal immunoglobulins. Antibody response to immunization antigens is abolished.
Eosinophilia may be observed in SCID, and IgE serum levels may be elevated in spite of hypogammaglobulinemia. Cytopenias, caused by infections or marrow damage, may also be present. Autoimmune hemolytic anemia is frequent in PNP deficiency.47 Marrow abnormalities (dysplasia or aplasia) can be observed in ADA,77 PNP,78 Cernunnos/XLF,79,80 and LIG481 deficiencies.
The diagnosis of ADA and PNP deficiency is facilitated by the demonstration of increased levels of deoxyadenosine triphosphate and deoxyguanosine triphosphate, respectively, in red blood cells.
Differential diagnosis of SCID includes secondary forms of immunodeficiencies, especially HIV infection, congenital rubella, and CMV infections, severe malnutrition, marrow failure syndromes,82 and defects of vitamin B12 and folate metabolism.83,84
SCID is a medical emergency and is inevitably fatal if untreated. Confirmation of diagnosis by appropriate laboratory assays, referral to a tertiary care center, and aggressive treatment of infections should be immediately initiated in infants with possible SCID. High-dose intravenous sulfamethoxazole-trimethoprim (20 mg/kg) is effective in treating P. jirovecii pneumonia. CMV should be treated with ganciclovir and adenoviral infections should be treated with cidofovir. Infants who have received bacillus Calmette-Guérin (BCG) vaccination at birth should receive isoniazid and rifampicin, regardless of the presence of overt signs of mycobacteriosis. Administration of IVIG and antimicrobial prophylaxis are necessary to reduce the risk of infections. Parenteral nutrition may be necessary, especially if chronic diarrhea and failure to thrive are present.
Survival, however, is ultimately dependent on immune reconstitution. Allogeneic HSCT was first performed in 1968 in an infant with X-linked SCID,85 and is the treatment of choice. A multiinstitutional analysis of outcome of HSCT for SCID performed in North America in the period 2000 to 2009 demonstrates that survival following HSCT from a human leukocyte antigen (HLA)-identical sibling is as high as 97 percent.86 T-cell–depleted transplantation from haploidentical donors results in a 79 percent 5-year-survival rate if no conditioning regimen is used. Importantly, survival is as high as 94 percent, regardless of donor type, if the transplant is performed within the first 3.5 months of age.86 Moreover, 90 percent survival rate has been reported for older infants who received HSCT and did not have a prior history of infection.86 For patients who do not have a matched sibling donor, use of pretransplantation conditioning regimen increases the chance of achieving more robust B-cell reconstitution, but this benefit must be balanced against toxicity of chemotherapy.86 By contrast, HSCT from unrelated cord blood is associated with a lower survival rate (58 percent).86
Failure to achieve sufficient T- and B-cell reconstitution is associated with prolonged morbidity after transplantation, but most patients with SCID enjoy good quality of life after transplantation.56 However, neurologic complications and developmental problems after transplantation are more common in patients with SCID associated with increased radiosensitivity and in patients with defects of purine metabolism.56,87,88,89
Enzyme replacement therapy offers rapid normalization of the toxic metabolites in ADA deficiency in those who do not have a matched donor, and may result in immune reconstitution and significant clinical improvement in patients with ADA deficiency, although T-cell counts often remain low.45
Gene therapy is an attractive form of therapy for SCID patients who lack fully matched donors. Transplantation of gene-modified autologous hematopoietic stem cells (aHSCs) may lead to immune reconstitution without the risk of graft-versus-host disease. More than 40 patients with ADA deficiency have received gene therapy using nonmyeloablative conditioning regimen and transfusion of aHSCs transduced with ADA-encoding retroviral vectors. All of these patients are alive, and approximately 75 percent of them have attained sufficient immune reconstitution.90,91,92
Twenty patients with X-linked SCID received gene therapy with retroviral vectors in Paris and London, without conditioning regimen. Seventeen of them are alive (as of this writing) with robust T-cell immune reconstitution, but variable B-cell function.93,94 However, five patients developed leukemia as the result of insertional mutagenesis.95,96 This prompted development of novel, hopefully safer, vectors. A new multiinstitutional trial of gene therapy for X-linked SCID with a self-inactivating γ-retroviral vector is being conducted as of this writing. Of nine patients treated, eight are surviving, and seven have attained good T-cell reconstitution. No leukemic proliferations have been observed.97 Variable, but often poor, B-cell reconstitution has been reported after gene therapy for X-linked SCID without conditioning. To overcome this problem, a new trial based on use of a self-inactivating lentiviral trial and reduced intensity conditioning is under way at the National Institutes of Health as of this writing.
OTHER COMBINED IMMUNODEFICIENCIES
In some cases, significant impairment of T-cell immunity is associated with residual development and/or function of T lymphocytes. These conditions are also known as CID to distinguish them from SCID, in which both T-cell development and function are abrogated. The clinical features of CID overlap with SCID, but also include autoimmunity and/or inflammatory manifestations reflecting unbalanced immune homeostasis. CID is caused by two main mechanisms: (1) hypomorphic mutations in SCID-causing genes that allow for some T-cell development; and (2) genetic defects that affect late stages in T-cell development or peripheral T-cell function.
Originally described in 1965, Omenn syndrome is characterized by severe infections, associated with early onset diffuse rash or generalized erythroderma, alopecia, eosinophilia, lymphadenopathy, hepatosplenomegaly, hypoproteinemia with edema, and oligoclonal expansion of activated autologous T lymphocytes that infiltrate and damage target tissues.98,99
Various gene defects can cause this syndrome. Hypomorphic mutations in the RAG1 and RAG2 genes are most common,100 but virtually any gene defect that severely impairs, but does not abolish, T-cell development may cause the disease.101
Defects of immunologic tolerance have been implied in the pathophysiology of Omenn syndrome. Thymic expression of AIRE (autoimmune regulator), a transcription factor involved in presentation of self-antigens and negative selection of autoreactive thymocytes, is reduced.102 Impaired generation of natural regulatory T cells, and homeostatic proliferation of T lymphocytes in a lymphopenic environment, may also play a critical role in the pathophysiology of the disease.103
Laboratory investigations demonstrate that leukocytosis with eosinophilia and hypogammaglobulinemia are common findings, and that serum IgE is often elevated. The number of circulating T lymphocytes may vary, but they have a characteristic activated/memory (CD45R0+) phenotype. T cells have a restricted repertoire, and the distribution of CD4 and CD8 subsets is generally skewed. There is also a skewing to a T-helper (Th) type 2 (Th2) profile, with increased production of IL-4 and IL-5. The in vitro lymphocyte response to antigens is abrogated; responses to mitogens are variable, but in general are reduced.74,104 The number of circulating B and NK lymphocytes may vary, depending on the nature of the underlying genetic defect. Absence of invariant NK T cells has been reported in RAG-deficient Omenn syndrome.105
Differential diagnosis includes maternal T-cell engraftment in patients with SCID, complete atypical DiGeorge syndrome, CHARGE syndrome (coloboma of the eye, heart defects, atresia of the nasal choanae, retardation of growth and/or development, genital and/or urinary abnormalities, and ear abnormalities and deafness), immune dysregulation-polyendocrinopathy-enteropathy-X-linked (IPEX) syndrome and other conditions of neonatal erythroderma.106,107,108,109 Male infants with NEMO deficiency can also present with severe skin manifestations resembling Omenn syndrome.
In preparation for allogeneic HSCT, the only curative treatment available,56 patients require aggressive nutritional support, correction of hypoproteinemia, and treatment or prevention of infections with antibiotics, antifungals, and immunoglobulin replacement therapy. Immune suppression with steroids or cyclosporine is beneficial in controlling T-cell–mediated tissue damage.
TCR ligation promotes activation of the p56Lck kinase, which mediates phosphorylation of components of the CD3 complex. This allows recruitment and phosphorylation of the zeta-associated protein of 70 kDa (ZAP-70), activation of downstream signaling molecules, release of Ca2+ from intracellular endoplasmic reticulum stores, and initiation of Ca2+ influx. Mutations of lymphocyte-specific protein tyrosine kinase (LCK), ZAP-70, and of other TCR-associated signaling molecules (RHOH, MST1, IL-2–inducible T-cell kinase [ITK]) result in various forms of CID with dysfunctional T cells.110,111,112,113,114,115 Finally, PI3K, composed of a p110δ and a p85 subunit, is involved in generation of phosphatidylinositol 4,5-triphosphate (PIP3) and activation of mammalian target of rapamycin (mTOR) and AKT. Activating mutations of the PI3KD gene (encoding for the PI3K subunit p110δ) results in increased activation-induced cell death of T lymphocytes, and consequently immunodeficiency.29,30
Patients with these disorders present with early onset and severe infections. Warts, molluscum contagiosum, infections caused by herpes viruses, and a high risk of Epstein-Barr virus (EBV)-driven lymphoproliferative disease have been reported in patients with LCK, RHOH, and MST1 deficiency, and with activating PI3KD mutations.29,30,110,113,114 Moreover, autoimmunity and lung granulomatous disease may also occur. From a laboratory standpoint, selective loss of CD8+ lymphocytes is observed in patients with ZAP-70 deficiency, and although the number of CD4+ lymphocytes is preserved, in vitro response to mitogens is markedly reduced, consistent with a signaling defect.111,112 Patients with LCK, RHOH, MST1, and ITK deficiency and with gain-of-function PI3KD mutations have a reduced number of naïve CD4+ T cells. Oligoclonality of the T-cell repertoire and an increased proportion of exhausted CD8+ T memory (TEMRA) cells have been reported in these patients.29,30,107,113,114,115
Differential diagnosis of ZAP-70 deficiency includes MHC class I deficiency and CD8α deficiency, two conditions characterized by a severe reduction of CD8+ lymphocytes. Patients with CD8α deficiency have an unusual population of CD3+ TCR αβ+ CD4– CD8– cells that have normal proliferative responses and usually survive to adulthood, although a late death from infections has been reported.116,117 The other defects of TCR signaling have an overlapping phenotype. Ultimately, biochemical and molecular tests are needed to define the diagnosis.
The only curative treatment of this group of disorders is allogeneic HSCT. Treatment with rapamycin (an mTOR inhibitor) or phosphoinositide 3-kinase inhibitor may reduce lymphoproliferation and hepatosplenomegaly in patients with activating PI3KD mutations.29,30
Following TCR signaling, the complex composed of MALT1, BCL-10, and caspase recruitment domain-containing protein (CARD)-11 proteins is activated, resulting in recruitment of TRIF-related adaptor molecule (TRAF) 6 and activation of IKK, permitting nuclear translocation of the p50 and p65 subunits of NF-κB and consequently induction of activation of NF-κB–dependent genes. Mutations of MALT1,118 CARD11,119,120 and IKBKB121 (encoding for the IKKβ component of the IKK complex) genes are associated with increased susceptibility to bacterial, viral and fungal infections. Although the number of circulating T lymphocytes is normal, generation of memory T cells is impaired and proliferative responses to CD3 stimulation are decreased. Patients with CARD11 mutations have a block in B-cell development at the transitional stage,119,120 and virtual absence of class-switched memory B cells is observed in patients with IKBKB mutations.121
As reported above, mutations of the IKBKG/NEMO gene are responsible for X-linked immunodeficiency with ectodermal dysplasia, whose clinical manifestations may also include opportunistic infections, resembling CID. Finally, gain-of-functions mutations of the IKBA gene, that prevent phosphorylation and degradation of the IKB-α subunit of the IKB complex, cause T-cell immunodeficiency with ectodermal dystrophy. The immunologic phenotype includes deficiency of memory T cells, impaired in vitro proliferation of naïve T cells to TCR/CD3 stimulation, and hypogammaglobulinemia with inability to mount specific antibody responses.122 In addition to TCR signaling, activation of toll-like receptor (TLR) and tumor necrosis factor (TNF) pathways can also be compromised, causing increased susceptibility to a broad range of pathogens (pyogenic bacteria, mycobacteria, Candida, other opportunistic pathogens).123
Coronin-1A is an actin regulator that is predominantly expressed in hematopoietic cells, plays a key role in regulating T-cell survival and migration. Mutations affecting both alleles of the CORO1A gene have been reported in patients with CID and an increased risk of severe varicella and EBV lymphoproliferative disease.124,125 The immunologic phenotype includes naïve T-cell lymphopenia with normal numbers of B and NK cells, oligoclonal T-cell repertoire and reduced number of circulating invariant NKT (iNKT) cells and mucosa-associated invariant T (MAIT) lymphocytes. Immunoglobulin serum levels are low, and antibody responses to antigens are absent. The disease can be treated by allogeneic HSCT.124
The CD27 costimulatory molecule regulates survival and activation of T, B, and NK cells. CD27 deficiency is a CID with risk of EBV lymphoproliferative disease. In vitro T-cell proliferation to mitogens and antigens is reduced.126 Immunoglobulin serum levels may be initially high, but patients eventually become hypogammaglobulinemic.
Cytidine 5-triphosphate synthase 1 (CTPS1) is involved in de novo synthesis of cytidine 5-triphosphate (CTP), a nucleotide required for DNA and RNA metabolism. Impaired de novo synthesis of CTP causes a proliferation defect in both T and B lymphocytes. CTPS1 mutations have been identified in several infants from Northwestern England. The disease is characterized by severe bacterial and viral infections since early in life, and an increased risk of EBV-driven non-Hodgkin lymphoma. There are no extra-immune manifestations. Variable degrees of lymphopenia (especially of CD4+ cells), increased proportion of effector memory T cells and reduced in vitro proliferation to mitogens and antigens have been reported. Immunoglobulin levels may be normal, but specific antibody titers are reduced, and there is a low number of memory B cells.127
MHC class I deficiency is characterized by reduced expression of MHC class I molecules at the cell surface. The disease is inherited as an autosomal recessive trait, and may be caused by defects in the TAP1,128 TAP2,129 or Tapasin130 genes. These defects interfere with intracellular transport of peptide antigens and their loading onto MHC class I molecules, and cell-surface expression of the complex.
MHC class I deficiency manifests with recurrent respiratory infections in childhood, and chronic inflammatory lung disease and skin lesions, mimicking Wegener granulomatosis in patients with transporter-associated with antigen-processing (TAP)-1 and TAP-2 deficiencies.131,132 Chronic lung disease is a prominent cause of death. Glomerulonephritis and herpes zoster infections have been reported in Tapasin deficiency.130
The number of circulating CD8+ T cells is reduced, because positive selection of CD8+ lymphocytes in the thymus depends on the recognition of MHC class I molecules. In vitro T-cell function is normal, which facilitates differential diagnosis with ZAP-70 deficiency in patients who have significantly reduced CD8+ cells. The NK cytolytic activity is usually significantly reduced (see Table 80–2). Serum immunoglobulin levels are variable.
Prophylactic measures, similar to those used in cystic fibrosis, may be beneficial. Treatment of the granulomatous lesions is based on use of topical antiseptics; immunosuppressive drugs may worsen symptoms and should be avoided.
MHC class II deficiency is defined by the lack of MHC class II expression and autosomal recessive inheritance. There is a higher prevalence in populations of North African origin. MHC class II deficiency is caused by mutations of transcription factors that bind to the proximal promoters of the MHC class II gene. Four different gene defects are known and include mutations of the CIITA, RFXANK, RFX5, and RFXAP genes.133
Typically, patients present early in life with increased susceptibility to bacterial, viral, and opportunistic infections. Severe lung infections, chronic diarrhea, and sclerosing cholangitis, often secondary to Cryptosporidium or CMV infection, are frequently observed. Less-severe presentations and survival into adulthood have been reported.133
The number of circulating CD4+ T cells is markedly reduced, reflecting an impairment of positive selection in the thymus. Delayed-type hypersensitivity responses are absent, but in vitro proliferative responses to mitogens are preserved. Hypogammaglobulinemia is common and poor antibody response to immunization antigens is consistently observed (see Table 80–2).133 The diagnosis is based on demonstrating lack of MHC class II expression on monocytes, B lymphocytes and in vitro activated T cells. Differential diagnoses include HIV infection and idiopathic CD4 lymphopenia; however, in these conditions expression of MHC class II molecules is preserved.
MHC class II deficiency has a poor prognosis. If untreated, most patients die in infancy or childhood. Respiratory infections are the predominant cause of death. Liver failure is observed in patients who develop sclerosing cholangitis. Antibiotic prophylaxis and immunoglobulin replacement therapy, with adequate nutritional support, are required. HSCT is the only curative approach, but survival rate is lower than in other forms of CID and graft-versus-host disease is common, especially in patients with preexisting viral infections.133
Calcium mobilization is a key event in the activation process of lymphocytes and nonimmune cells. Two molecules, calcium release-activated calcium channel protein 1 (ORAI1) and stromal interaction molecule 1 (STIM1), mediate the function of Ca2+ entry channels. ORAI1 is a ubiquitously expressed protein that constitutes the pore-forming subunits of the Ca2+ release-activated channels located in the cell membrane. STIM1 senses the Ca2+ concentration in the endoplasmic reticulum and activates Ca2+ release-activated channels. Mutations of both the ORAI1 and STIM1 genes in humans result in an autosomal recessive immunodeficiency with increased susceptibility to severe infections, especially from herpesviruses infections, associated with nonprogressive myopathy and ectodermal dysplasia. Manifestations of immune dysregulation (autoimmune cytopenias, hepatosplenomegaly) are common, especially in STIM1 deficiency.134,135 Although T-cell development is unaffected, in vitro
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


