PROMISE OF IMMUNOTHERAPY
The ability of the immune system to recognize and eradicate cancer was first postulated in the 19th century; however, proof of principle remained elusive until the 20th century. The effect of infection on tumor regression was observed as early as 1884 by Anton Chekov (1); following this, William Coley developed a mixture of killed bacteria that were used to treat various types of cancer between the 1890s and 1960s with mixed clinical benefit. In addition, the concept of using bacterial elements was validated with Food and Drug Administration (FDA) approval of intravesical administration of BCG, which is an FDA-approved therapy that leads to nonspecific inflammatory immune responses and clinical benefit for patients with superficial bladder cancer (2). The discovery of the major histocompatibility complex (MHC) and T-cell receptor (TCR) in the 1980s provided insight into T-cell function that led to a number of clinical trials (3,4). Unfortunately, many of the early clinical trials failed due to incomplete understanding of T-cell function. Further research led to understanding of costimulatory and coinhibitory molecules, which led to improved strategies in the field of cancer immunotherapy, including chimeric antigen receptor (CAR) T cells and immune checkpoint therapies, resulting in clinical success that turned the tide in favor of immunotherapy.
The basic principles that guide cancer immunology are (a) immune surveillance, (b) immune editing, and (c) immune tolerance (5). Immune surveillance involves scanning and eliminating the transformed nascent cells by the immune system. This theory was bolstered by the finding of tumor-specific antigen that could be identified by cytotoxic T cells (6). Immune editing is a process that involves elimination in which the immune system acts as an extrinsic tumor suppressor (similar to the original concept of immunosurveillance); second, equilibrium occurs, in which tumor cells survive but are held in check by the immune system; and third, in escape, tumor cell variants with either reduced immunogenicity or the capacity to attenuate or subvert immune responses grow into clinically apparent cancers. Escape theory also gave credence to the concept of immune tolerance by which cancer cells exploit the body’s immune system and use it for their continuous immune evasion and subsequent growth and proliferation.
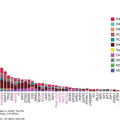
The immune compartments that aid in tumor elimination and promote tumor evasion are listed in Table 48-1. The guiding theory for development of immunotherapy is to promote antitumor immune factors and attenuate protumor factors.
| Antitumor | Protumor | |
|---|---|---|
Innate: Mature dendritic cell Tumor-associated macrophage (M1 phenotype) Tumor-associated neutrophil (N1) | Innate: Immature dendritic cell Tumor-associated macrophage (M2 phenotype) Tumor-associated neutrophil (N2) | |
| Cellular compartment | NK cells Adaptive: CD8+ T cells CD4+ T cells: Th-1, Th-9, Th-17 |
Adaptive: CD4+ T cells: Th-9, Th-17 T – regulatory cells |
| Soluble factors (cytokines) | Granzyme B, IL-1 a,b, IL-2, IL-6, IFN-g, IL-12, IL-17 | TGF-b, IL-2, IL-4, IL-6, IL-10, IL-17, IL-23 |
CELL-BASED THERAPY
Immune cells, such as cytotoxic T cells, are one of the main effector mechanisms of tumor killing and elimination. Cell-based therapy involves increasing the number and efficiency of immune cells that are capable of recognizing and killing the tumor cells
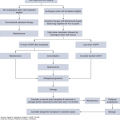
Adoptive T-cell therapy (ACT) involves adoptive transfer of autologous tumor-infiltrating lymphocytes (TILs) to the tumor-bearing host (7,8). It involves isolation and ex vivo rapid expansion of TILs from tumor, followed by infusion of large numbers of expanded autologous TILs as depicted in Fig. 48-1. Therapy with TILs has been especially promising for patients with stage III and IV unresectable melanoma, and its efficacy has been further increased with combined interleukin (IL) 2 therapy and preconditioning of the host with chemotherapy agents such as Cytoxan and fludaribine. Objective response has been reported in more than 50% of patients (8,9). The transition of ACT into the clinical setting, however, has not been without difficulties. These can be considered in two groups: factors relating to difficulties in generating appropriate products for adoptive transfer and factors relating to host or tumor resistance to transferred populations. Recent advances in development of artificial antigen-presenting cells (aAPCs) have helped to overcome many technical issues with rapid TIL expansion (10). Further, TIL therapy has been shown to be particularly beneficial in patients pretreated with other immunotherapy agents such as IL-2 or checkpoint blockade like anti-CTLA4 (anti–cytotoxic T lymphocyte-associated antigen 4) (8).
FIGURE 48-1
Adoptive T-cell therapy. There are currently two major sources of T cells used in adoptive cell therapy for melanoma and other cancers. A. The first form of therapy uses tumor-infiltrating lymphocytes (TILs) expanded ex vivo from surgically resected metastatic tumors. The TILs are initially expanded ex vivo using anti-CD3 activation in the presence of irradiated PBMC feeder cells and IL-2. The final product is infused into the patient. B. The second major approach for adoptive T-cell therapy uses T cells expanded from autologous PBMCs that undergo one of three possible manipulations to enrich the cells for tumor antigen-specific T cells.
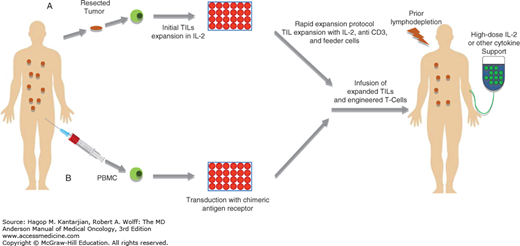
In an effort to increase the affinity of T cells to tumor antigen, CAR T cells are engineered that contain a synthetic antigen receptor with specificity to a tumor cell marker coupled with T-cell signaling domain, resulting in high-affinity recognition of the tumor antigen (11). The CAR T cells are particularly effective against cancer with known tumor antigen. The CAR T cells with specificity for CD19 have shown activity in chronic lymphocytic lymphoma (12). Recently, a phase III study demonstrated the CD19-coupled CAR T cells were particularly effective against relapsed refractory acute lymphoblastic leukemia (ALL) (13,14). Autologous T cells transduced with lentiviral vector expressing a chimeric antigen receptor with specificity for the B-cell antigen CD19, coupled with CD137/4-1 BB (a costimulatory receptor in T cells) and CD3-zeta (a signal transduction component of the T-cell antigen receptor) signaling domains were infused in patients with relapsed or refractory ALL and resulted in complete remission in 90% of cases. A potential drawback of CAR T-cell therapy is exaggerated immune response against normal tissue expressing the antigen; therefore, careful dose escalation studies are necessary to predict the therapeutic window. Further, in many cancers the tumor antigen is not known, making CAR T-cell therapy ineffective against undefined tumor antigens.
CANCER VACCINE
Tumors express a vast array of antigens. Cancer vaccines typically consist of a known tumor antigen with an immunostimulatory formulation that could activate the tumor antigen-specific lymphocyte population, leading to eradication of cells expressing this antigen.
Although therapeutic cancer vaccines have impressive antitumor activity in various animal models, their clinical benefit in cancer patients has to date been minimal (15). However, optimization of a cancer vaccine with a combinatorial strategy resulted in significant success of the cancer vaccine, most notably two prostate cancer vaccines: Provenge and Prostvac (16,17). Provenge (sipuleucel-T) is a cancer vaccine based on dendritic cells (DCs) that was approved in 2010. Provenge is prepared from patient’s peripheral blood mononuclear cells (PBMCs) pulsed with a fusion protein of granulocyte-macrophage colony-stimulating factor (GM-CSF) and the prostate cancer antigen prostatic acid phosphatase (PAP), with the rationale that on injection GM-CSF–activated DCs will present the antigen (PAP) to T cells in a more efficient manner. Provenge increased the median survival by 4.1 months compared to placebo in metastatic prostate cancer. Prostvac is another prostate cancer vaccine that is composed of vaccinia and fowlpox vector that contain the transgenes for prostate-specific antigen (PSA) and multiple T-cell costimulatory molecules (TRICOM) (18). The TRICOM consists of costimulatory molecules B7.1, leukocyte function-associated antigen 3 (LFA-3), and intercellular adhesion molecule 1 (ICAM-1). The PSA-TRICOM vaccines infect antigen-presenting cells (APCs) and generate proteins that are expressed on the surface of APCs that present antigen to T cells, resulting in antigen-specific tumor killing. E75 (nelipepimut-S) is a human leukocyte antigen (HLA) A2/A3–restricted immunogenic peptide derived from the HER2 protein, which was used in a phase I/II trial involving patients with node-positive or high-risk node-negative breast cancer expressing any level of Her-2 to prevent recurrence (19).
Although there remains significant room for improvement in the design and delivery of cancer vaccines, these clinical successes have led to the proof of principle that cancer vaccines are effective. Few of the cancer vaccines that are approved or are currently in clinical trials are listed in Table 48-2.
| Types of Cancer | Target Antigen | Formulation |
|---|---|---|
| Breast | Her-2/Neu | E75, a human leukocyte antigen (HLA) A2/A3-restricted HER-2/neu (HER2) peptide and granulocyte-macrophage colony-stimulating factor. |
| Breast | Mammoglobin-A | A plasmid encoding the mammaglobin-A gene with potential immunostimulating and antineoplastic activities |
| Prostate | PAP | Autologous PBMC loaded with GM-CSF+PAP fusion protein |
| Prostate | PSA | Vaccinia and fowlpox virus encoding PSA and CD86, ICAM-1, and LFA3 |
| Follicular B cell Lymphoma | Idiotype |