Iatrogenic Hypoglycemia
Stephanie Anne Amiel
No definition of good glycemic control in diabetes is complete unless it includes a statement about absence of hypoglycemia. Patients with diabetes and their families are well aware of this, and healthcare professionals responsible for patients with diabetes need to be equally well informed.
Hypoglycemia—literally, a low blood glucose concentration—is rarely encountered in healthy individuals. Glucose is normally the major metabolic fuel for the brain. Because the brain stores only trivial amounts of glucose as glycogen, the brain is dependent for normal function on an adequate supply of glucose from its circulation. Both the entry of glucose into the circulation and its removal therefrom vary widely with fasting and feeding and with rest and exertion. Elaborate and sensitive mechanisms exist to maintain glucose concentrations in the plasma within narrow limits—too high a concentration upsets the water balance in tissues, causes glycosuria, and accelerates tissue glycosylation, whereas too low a concentration results in cerebral dysfunction and ultimately coma and death. In health, hypoglycemia sufficient to cause clinically significant cognitive dysfunction does not occur because of the efficiency of the endogenous mechanisms of glucose homeostasis. Once these mechanisms are upset by diabetes and its treatment, clinically problematic hypoglycemia can occur.
Hypoglycemia can be defined in biochemical terms as a plasma glucose concentration below the normal range. It should be noted that glucose concentrations in plasma are approximately 10% higher than those in whole blood because red cells contain relatively low concentrations of glucose; arterial glucose levels are higher than venous levels, and capillary levels are between the arterial and venous levels. Research studies of hypoglycemia in human subjects often use arterialized venous glucose concentrations because they approximate those of the arterial system (and therefore the glucose supply to a tissue) and make arterial sampling unnecessary (1).
Fasting plasma glucose concentrations are considered normal if they are less than 6 mmol/L, although levels higher than 5.5 mmol/L are rarely seen in healthy individuals (2). The lower limit of normality is more equivocal. Healthy people rarely have a plasma glucose concentration lower than 4 mmol/L after an overnight fast, but levels of 2.8 mmol/L have been recorded in healthy people after prolonged fasting (3). Spontaneous, pathologic hypoglycemia, such as may occur in the presence of
insulin- or insulin-like growth factor (IGF)-secreting tumors, requires for diagnosis the demonstration of plasma glucose levels of less than 2.8 mmol/L or even, according to some authorities, levels less than 2.2 mmol/L in women (4). However, detailed physiologic studies show a detectable impairment of higher cerebral function at plasma glucose levels of 3 mmol/L (arterialized venous plasma) (5), and there is evidence that recurrent plasma glucose levels of 3 mmol/L damage the endogenous protective mechanisms against more severe hypoglycemia (6). Nonpancreatic counterregulatory responses to hypoglycemia start at 3.5 to 3.6 mmol/L (see below). Thus, in the context of the treatment of diabetes mellitus, hypoglycemia is defined as a plasma glucose concentration of 3.5 mmol/L or less.
insulin- or insulin-like growth factor (IGF)-secreting tumors, requires for diagnosis the demonstration of plasma glucose levels of less than 2.8 mmol/L or even, according to some authorities, levels less than 2.2 mmol/L in women (4). However, detailed physiologic studies show a detectable impairment of higher cerebral function at plasma glucose levels of 3 mmol/L (arterialized venous plasma) (5), and there is evidence that recurrent plasma glucose levels of 3 mmol/L damage the endogenous protective mechanisms against more severe hypoglycemia (6). Nonpancreatic counterregulatory responses to hypoglycemia start at 3.5 to 3.6 mmol/L (see below). Thus, in the context of the treatment of diabetes mellitus, hypoglycemia is defined as a plasma glucose concentration of 3.5 mmol/L or less.
In diabetes, hypoglycemia is also often defined by its clinical presentation. Acute hypoglycemia is symptomatic, and Whipple’s triad, although defined by a surgeon interested in insulinomas, remains a useful guide for defining episodes. The triad requires (a) symptoms attributable to a low plasma glucose concentration, (b) a measurably low plasma glucose concentration (less than 3.5 mmol/L in diabetes), and (c) rapid resolution of the symptoms after correction of the biochemical abnormality (7). However, as will be described later, patients with diabetes (and indeed those with insulinomas) can lose their ability to generate and detect the symptoms of early hypoglycemia (see Hypoglycemia Unawareness), and Whipple’s original triad is best modified by the inclusion of the term “and signs” in the first criterion (Table 40.1).
TABLE 40.1. A Modification of Whipple’s Triad for Diagnosis of Hypoglycemia in Diabetes Therapy | |||
|---|---|---|---|
|
Perhaps the most important clinical definition of acute hypoglycemia in diabetes therapy is the division into mild and severe. Many authorities also recognize an intermediate category of moderate. Mild hypoglycemia is defined as that which is recognized and treated by the patient, and severe hypoglycemia is defined as that which the patient is unable to self-treat because of cognitive impairment. Some authorities restrict the term “severe hypoglycemia” to episodes requiring parenteral therapy (intramuscular glucagon or intravenous glucose) and/or those resulting in coma or seizure. The classification “moderate hypoglycemia” usually refers to episodes that the patient could self-treat but resulted in significant life disruption. The lack of clarity in this definition has led to its disuse. The terms “mild” and “severe” hypoglycemia are not appropriately applied to degrees of biochemical hypoglycemia (Table 40.2).
TABLE 40.2. Clinical Classification of Acute Hypoglycemia | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
FREQUENCY OF HYPOGLYCEMIA
Formal studies of the frequency of hypoglycemic events must be compared carefully because of differences in definitions. This difficulty not withstanding, it is important to document the frequencies of hypoglycemic episodes in order to compare the effects of different therapeutic regimens and to determine clinical characteristics that put patients at risk.
Mild hypoglycemia, although often unpleasant, tends to be accepted as an inevitable consequence of glucose-lowering therapies. However, mild hypoglycemia should not be ignored, as it has the potential to increase the risk of severe episodes. In the Diabetes Control and Complications Trial (DCCT), which was conducted in patients with type 1 diabetes, the rate of severe hypoglycemia increased from approximately 20 episodes per 100 patient-years during conventional therapy to 60 per patient-year during intensified therapy (8). In contrast, the Dusseldorf group, using internally consistent definitions but different from those used in the DCCT, reported a decline in rates of severe hypoglycemia with intensified therapy from rates of 28 to 17 episodes per 100 patient-years (9).
COUNTERREGULATION: THE DAMAGED DEFENSES AGAINST SEVERE HYPOGLYCEMIA IN DIABETES
As the plasma glucose concentration begins to decrease, an orchestrated neurohumoral response acts to prevent hypoglycemia. The first component of this counterregulation is local to the pancreatic islets, with cessation of insulin secretion and stimulation of glucagon release by the pancreas. These responses promote hepatic production of glucose, which, in health, represents the most important mechanism for preventing hypoglycemia between meals. In people with diabetes, the insulin levels do not decrease as glucose levels fall, either because of persistent absorption of exogenous insulin or because of non-glucose-dependent elevation of insulin secretion by sulfonylureas. The lack of decline in plasma insulin concentration as glucose levels decrease then results in iatrogenic hypoglycemia.
In addition to their inability to modulate insulin levels as the plasma level of glucose decreases, patients with type 1 diabetes lose the ability to enhance glucagon production by pancreatic α-cells in response to hypoglycemia. Basal glucagon production and glucagon responses to other secretagogues are maintained, but the response to hypoglycemia is abrogated. This happens within 5 years of the onset of type 1 diabetes (10) and may be secondary to decreased β-cell activity. Glucagon responses are also lost late in the evolution of type 2 diabetes, and the glucagon response to hypoglycemia is reduced under experimental conditions in which β-cell activity is maintained by sulfonylureas (11,12).
In experimental studies, counterregulation (the spontaneous correction of a decreasing plasma glucose level) occurs even if the insulin and glucagon responses are inhibited (13), a consequence of secondary defense mechanisms involving the autonomic nervous system and adrenal medulla. Released epinephrine and norepinephrine stimulate endogenous glucose production and also inhibit glucose consumption by peripheral tissues. These actions occur through elevation of substrates for gluconeogenesis, including nonesterified fatty acids, which also inhibit peripheral glucose oxidation (14). Glucose is generated initially by hepatic glycogenolysis and subsequently by hepatic (and renal) gluconeogenesis (15,16). The activation of the sympathetic
nervous system can be demonstrated not only by determining adrenal catecholamine secretion as blood glucose concentrations fall but also by determining sympathetic activity in muscle (17), recording perspiration (18), or measuring norepinephrine assumed to have “spilled over” from sympathetic nerve terminals.
nervous system can be demonstrated not only by determining adrenal catecholamine secretion as blood glucose concentrations fall but also by determining sympathetic activity in muscle (17), recording perspiration (18), or measuring norepinephrine assumed to have “spilled over” from sympathetic nerve terminals.
A significant and probably growing number of patients with type 1 diabetes develop defects in the adrenal and autonomic responses to hypoglycemia. These occur independently of classical autonomic neuropathy, to which they are not mechanistically related (19) and represent an important risk factor for susceptibility to severe hypoglycemia. The defects are associated with a failure to generate the typical symptoms of autonomic activation. Affected patients, described as having hypoglycemia unawareness, are at greatly increased risk for severe hypoglycemia (20,21).
Growth hormone and cortisol are additional hormones with important roles in glucose homeostasis, and their absence can be associated with clinical hypoglycemia. Their roles in the response to acute hypoglycemia are less clear, and they certainly cannot compensate for defects in epinephrine secretion and autonomic responsiveness.
SYMPTOMS OF ACUTE HYPOGLYCEMIA
Early experts in diabetes, America’s Elliot P. Joslin and Britain’s R. D. Lawrence (22,23), divided the symptoms of acute hypoglycemia on physiologic principles into autonomic (attributable to activation of the autonomic nervous system and adrenal medulla) and neuroglycopenic categories (related to impaired glucose supply to the cerebral cortex). More recently, complex statistical packages have been used to group the symptoms reported by patients (or symptoms and signs reported by parents of children with diabetes) into related “families,” with very similar results (Tables 40.3 and 40.4) (24,25). Clinically, the specific symptoms experienced are not important as long as the patient becomes aware of them at a glucose level when cognitive function is adequate to enable an effective response (i.e., ingestion of some rapidly absorbed carbohydrate). Many patients with type 1 diabetes report a greater dependence on neuroglycopenic symptoms than on autonomic symptoms over time.
TABLE 40.3. Classification of Acute Hypoglycemia by Factor Analysis in Adults | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
TABLE 40.4. Classification of Acute Hypoglycemia by Factor Analysis in Children | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
The etiology of hunger, which represents a frequent symptom of hypoglycemia and most directly encourages resuscitative food intake, has not been clearly delineated. The satiety centers lateral to the hypothalamus may be involved. Leptin does not respond acutely in hypoglycemia, and the role of orexin is uncertain (26,27).
INVESTIGATION OF RESPONSES TO ACUTE HYPOGLYCEMIA
The categorization of the symptoms of acute hypoglycemia discussed above was based on data collected from the description of symptoms and signs by patients and their family members. More detailed studies on the relationships between symptoms of acute hypoglycemia and the effect of diabetes and its therapy often have involved experimentally induced hypoglycemia. Insulin injection (i.e., the insulin tolerance test) is a high-risk procedure that results in an uncontrolled, rapid, variable, and often profound hypoglycemic challenge that will miss all but the most extreme variations in the counterregulatory response. A slow decline in plasma glucose level induced by a low-dose infusion can be useful for examining an individual’s overall counterregulatory capacity (i.e., the ability to arrest or reverse a drop in glucose level) (28). However, the comparison of responses between individuals or groups of individuals may be complicated by the failure to achieve equal levels of hypoglycemia. As an alternative, controlled hypoglycemia can be induced with a modification of the euglycemic insulin clamp technique (29). In the slow-fall clamp, plasma glucose is controlled during a high-dose insulin infusion by means of an adjustable intravenous glucose infusion (Fig. 40.1). For patients with diabetes, plasma glucose often is regulated during the night preceding the study by an intravenous insulin infusion. This ensures that all subjects in a given study, with or without diabetes, are exposed to the same hypoglycemic challenge (i.e., similar initial glucose level, final glucose level, and rate of decline) (29). In some experimental protocols, the plasma glucose concentration is lowered in a series of steps, so that the glucose level associated with the onset of any one response can be determined for that individual or group of individuals. This technique first was used to determine the effect of the rate of glucose decline on the magnitude of the counterregulatory responses to a given moderate hypoglycemic challenge (29) and later to investigate the effects of intensified diabetes treatment on counterregulatory hormone responses (30). It is important to realize that the clamp technique does not examine
counterregulation per se, because the glucose level is always under the control of the investigator.
counterregulation per se, because the glucose level is always under the control of the investigator.
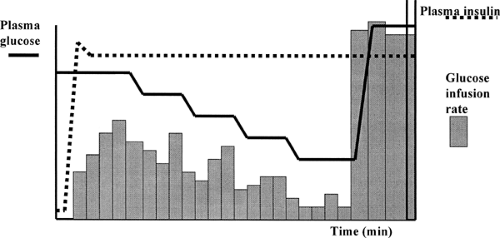 Figure 40.1. The stepped hypoglycemic clamp: a means of applying a controlled reproducible hypoglycemic challenge. A primed-continuous intravenous insulin infusion is started at time = 0 minutes, creating a predictable rapid square wave rise in circulating insulin levels (black dotted line). The subsequent hypoglycemia is controlled by a variable-rate glucose infusion (bars) adjusted according to the plasma glucose measurement made at the bedside every 5 minutes. (From Amiel SA, Simonson DC, Tamborlane WV, et al. Rate of glucose fall does not affect counterregulatory hormone responses to hypoglycemia in normal and diabetic humans. Diabetes 1987;36:518–522.) |
Such studies show that acute hypoglycemia results in a hierarchy of responses. In healthy volunteers, epinephrine secretion begins at arterialized plasma glucose levels of 3.0 to 3.4 mmol/L, adrenergic symptoms at 3.2 mmol/L, and minor but detectable cognitive impairment (first shown as a slowing of complex reaction times) at 3.0 mmol/L (Fig. 40.2) (29,30,31,31,32,33). More extensive cognitive impairment has been shown at lower glucose levels (32). The hormonal and symptomatic responses occur at higher glucose levels in patients with poorly controlled diabetes (31,34). The initiation of hormonal responses, and especially of symptoms, before the onset of significant cognitive impairment during a progressive glucose decline is critical to a patient’s ability to prevent severe hypoglycemia, giving a window of opportunity for recognition of the situation and for self-treatment (35).
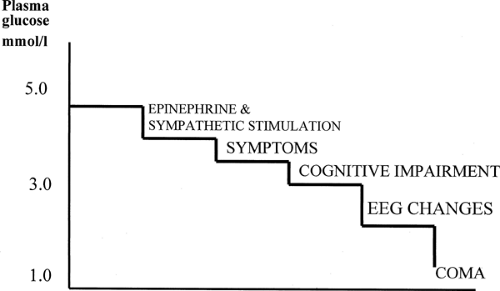 Figure 40.2. Normal hierarchy of responses to acute hypoglycemia. As plasma glucose level falls, the autonomic and symptomatic responses precede the onset of even mild cognitive impairment. |
The delineation of cognitive impairment during acute hypoglycemia is less clear. The brain is not a homogeneous organ, and specific brain functions use various brain regions that have different susceptibilities to hypoglycemia. The detection of hypoglycemia by glucose-sensing neurons and the initiation of the autonomic responses normally occur at or before the onset of cognitive changes (typically at arterialized plasma glucose levels of 3.4 mmol/L). For example, the slowing of choice reaction time occurs at glucose levels of 3 mmol/L, and the slowing of a simple motor activity such as finger tapping occurs at 2.4 mmol/L (Fig. 40.3).
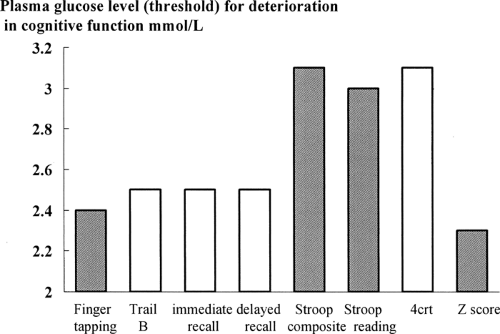 Figure 40.3. Glucose thresholds for onset of cognitive impairment in healthy volunteers. The data are acquired from clamp studies in which the rate of glucose decline is precisely controlled. It is important to note that different cognitive functions have different susceptibilities to hypoglycemia. Data for the two memory tests (immediate and delayed recall) and the composite Stroop score are from Cryer PE, Scott A, Segal MD, et al. Blood brain glucose transport is not increased following hypoglycemia. Diabetes 2000;39[Suppl 1]; the z scores are from Fanelli CG, Epifano L, Rambotti AM, et al. Meticulous prevention of hypoglycemia normalizes the glycemic thresholds and magnitude of most of neuroendocrine responses to, symptoms of, and cognitive function during hypoglycemia in intensively treated patients with short-term IDDM. Diabetes 1993;42:1683–1689; and the finger tapping, trail B, Stroop reading, and 4-choice reaction time (4crt) are from Hopkins DFC, Evans M, Lomas J, et al. Effects of antecedent control on cognitive function and symptoms during hypoglycaemia in type 1 diabetes. Diabet Med 1998;15[Suppl]:S4. The shaded bars represent cognitive functions that have been shown to manifest a degree of resistance to hypoglycemia in patients with hypoglycemia unawareness. |
HYPOGLYCEMIA UNAWARENESS
The best defense a patient with diabetes has against severe hypoglycemia is subjective awareness of an early fall in plasma glucose concentrations and the immediate ingestion of a rapidly available carbohydrate. Loss of the ability to generate and/or perceive such symptoms—the syndrome of hypoglycemia unawareness—is a serious clinical problem, increasing the risk of severe hypoglycemia by about 10-fold (20,21). Such unawareness places the patient in danger, as he or she may experience blood glucose levels low enough to impair basic cognitive processes such as reaction times (5), making common tasks such as driving or operating heavy machinery potentially lethal. Less dramatic, but equally devastating, is the sudden inability to sustain logical thought or the sudden onset of uncharacteristic aggression, which can have serious professional and social consequences for the individual. Because, by definition, a patient with hypoglycemia unawareness cannot recognize his or her state, a full assessment of a patient’s experience of hypoglycemia can be completed only by interviewing the people close to the patient in everyday life: the partner, other family members, or close friends (36).
An impaired ability to decrease insulin levels in response to hypoglycemia is integral to treatment with insulin or a sulfonylurea. Failure of the glucagon response in type 1 diabetes (and late insulin-deficient type 2) is apparently universal, and early studies also showed a marked diminution of the epinephrine response to hypoglycemia in patients with long-standing (10 years or more) type 1 diabetes (10). However, most patients with long-standing type 1 diabetes do retain an ability to recognize early hypoglycemia and defend against severe episodes, at least most of the time. Loss of awareness of hypoglycemia is certainly more common in long-duration type 1 diabetes than in diabetes of shorter duration (it has not been well described in type 2 diabetes), with 25% of patients in one general diabetes clinic with a duration of diabetes of more than 15 years reporting this problem. Such patients are at much higher risk of severe hypoglycemia (20,21). Similarly, more than a decade ago, patients with a history of recurrent severe hypoglycemia were found to have defective counterregulation, with impaired hormonal responses and an inability to arrest a glucose fall during low dose insulin infusion independent of diabetic neuropathy (19).
Interest in the problems of hypoglycemia awareness increased with the recognition that both asymptomatic (37) and severe symptomatic (8) hypoglycemia were more common in patients with type 1 diabetes randomized to receive intensified insulin therapy in the large trials of prevention of chronic complications (38). Investigation showed that patients receiving intensified therapy were less able than those receiving standard therapy to produce a counterregulatory response in the face of
insulin infusion (as were previously studied patients with recurrent hypoglycemia) (39). The defect was found to be a lowering of the plasma glucose level needed to initiate the adrenergic and symptomatic responses (30), closing, if not obliterating, the gap between the onset of symptomatic protective responses and of cognitive impairment (Figs. 40.3 and 40.4).
insulin infusion (as were previously studied patients with recurrent hypoglycemia) (39). The defect was found to be a lowering of the plasma glucose level needed to initiate the adrenergic and symptomatic responses (30), closing, if not obliterating, the gap between the onset of symptomatic protective responses and of cognitive impairment (Figs. 40.3 and 40.4).
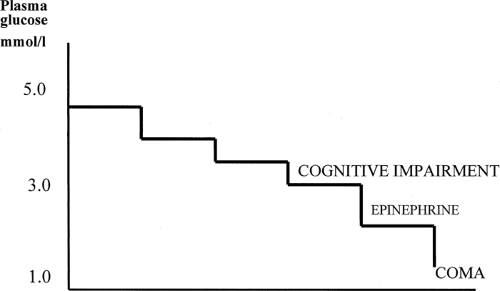 Figure 40.4. Disturbed hierarchy of responses to acute hypoglycemia after a preceding period of hypoglycemia. Counterregulatory failure is manifest as a lowering of the glucose level that induces an adrenergic counterregulatory response toward, or even below, that associated with the onset of cognitive impairment. |
There is little doubt that the detection of hypoglycemia and the initiation of the autonomic and adrenergic responses are centrally mediated. Animal studies show that maintenance of glucose supplies in a brain region around the ventromedial hypothalamus during systemic hypoglycemia prevents the counterregulatory response (40) and, conversely, that localized intracellular glucose deprivation (by deoxyglucose) in that brain region alone can induce a peripheral hyperglycemic “counterregulatory” hormonal response in euglycemic animals (41). Neurons from these and related brain areas can be excited by changes in glucose supply (42). In an experiment of nature, a patient with a sarcoid lesion in the same region showed abnormalities of glucose homeostasis and counterregulation until his lesion regressed with therapy (43). Glucose-sensing neurons also are present in the hepatic portal tract, as shown by animal experiments in which portal perfusion with glucose ameliorates the autonomic and adrenergic responses to systemic hypoglycemia (44,45), but their role in maintaining glucose supplies to the brain glucose is unclear. One hypothesis is that these neurons monitor glucose input from the gastrointestinal tract and may be able to moderate a centrally mediated counterregulatory response to systemic hypoglycemia once eating starts (46).
There is good circumstantial evidence that the trigger for autonomic and adrenergic counterregulation is a slowing of metabolic rate in the glucose-sensing neurons. Supplying the nonglucose substrates lactate or 3-hydroxybutyrate to support metabolism during experimentally induced hypoglycemia delays (i.e., shifts to lower glucose levels) the onset and magnitude of the epinephrine response, adrenergic symptoms, and cognitive impairment of acute hypoglycemia in volunteers (47,48,49). One popular current theory to explain the change in hypoglycemia sensitivity of the glucose-sensing mechanism is that an upregulation of glucose transporters and of glucose uptake by the brain occurs as a result of prior exposure to hypoglycemia (50,51,52). A study in patients with diabetes and healthy volunteers that calculated glucose consumption by the brain from measurements of cerebral blood flow and arteriovenous differences in glucose concentrations across the brain showed no effect of hypoglycemia in diabetic patients with a history of hypoglycemia unawareness, whereas hypoglycemia was associated with a decline in glucose uptake by the brain in symptomatic diabetic and nondiabetic subjects (53). A similar apparent adaptation in the handling of glucose by the brain also was induced in healthy volunteers by exposure to 56 hours of induced moderate hypoglycemia (54). The concept that the human brain can adapt to antecedent hypoglycemia by upregulating its ability to extract glucose and thus fails to trigger counterregulation because metabolism in the cerebral glucose sensor is better maintained is an attractive one. However, recent data obtained with positron emission tomography, measuring uptake in the brain of glucose labeled with a positron emitter, have been unable to confirm the data from the studies of arteriovenous difference (55,56). The discrepancies remain to be explained. One possibility is that adaptational changes are regional—all the described studies have measured only global glucose uptake by the brain. Regional differences in sensitivity to hypoglycemia itself have already been mentioned, and regional differences within the metabolic capacity of the brain and even of just the cerebral cortex have been suggested by further studies of nonglucose metabolic substrates in acute hypoglycemia (57).
The altered sensitivity of the brain’s hypoglycemia sensing mechanisms in the hypoglycemia unawareness syndrome is well documented. There is debate about the extent to which this effect occurs in the cerebral cortex. The discrepancies in the literature are almost certainly due to differences in the cognitive function tests used to assess cortical function during acute hypoglycemia—some of which adapt (i.e., do not show evidence of deterioration until a more profound hypoglycemia has been reached) and some of which do not. It is clear that, in people with counterregulatory failure and hypoglycemia unawareness, the glucose level required to initiate the symptomatic counterregulatory responses is reduced below normal to a greater extent than any change in glucose level associated with detectable cognitive malfunction, narrowing or even closing the window of opportunity between subjective awareness and confusion.
It is not known how antecedent hypoglycemia alters the response to subsequent episodes. As explained above, in connection with the mechanisms of hypoglycemia unawareness, a possible mechanism is upregulation of glucose transporters in
response to the initial hypoglycemic episodes; another is hypoglycemia-associated apoptosis of glucose-sensing cells (58). A suggested factor may be the adrenocorticotropic hormone or cortisol response to the initial insult (59,60), although how this might be involved in long-term hypoglycemia unawareness is unclear.
response to the initial hypoglycemic episodes; another is hypoglycemia-associated apoptosis of glucose-sensing cells (58). A suggested factor may be the adrenocorticotropic hormone or cortisol response to the initial insult (59,60), although how this might be involved in long-term hypoglycemia unawareness is unclear.
Whatever the mechanism, the link between the defective counterregulatory responses of hypoglycemia unawareness and hypoglycemia itself is strong. A series of research studies have demonstrated induction of counterregulatory failure and diminished symptomatic responses to experimentally induced hypoglycemia in both healthy volunteers and diabetic patients as a consequence of exposure to hypoglycemia during the day or night before the test stimulus (61,62,63,64,65). Such transient defects in the neuroendocrine response to hypoglycemia are very similar to those first demonstrated in intensively controlled diabetic patients with hypoglycemia unawareness: a lowering of the glucose level required to initiate the symptomatic and hormonal responses (particularly the epinephrine response) and a diminution of the hormonal response at any given glucose level. The relevance to the clinical syndrome of hypoglycemia unawareness in diabetic patients is confirmed by the demonstration that both the unawareness and the neurohumoral failure are either partially (66) or completely (67) reversible by scrupulous avoidance of hypoglycemia in daily living. It is important to note that the induction of temporary counterregulatory failure in experimental studies can be generated by modest prior decreases in plasma glucose concentrations (Table 40.5) and that reversibility in patients with diabetes can be achieved by successful avoidance of blood glucose concentrations lower than 3 mmol/L on home monitoring. This illustrates the danger of using definitions of hypoglycemia coined for the diagnosis of pathologic spontaneous hypoglycemia in the context of diabetes therapies.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


