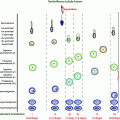
Fig. 7.1
Adrenal steroid synthesis. StAR steroidogenic autoregulatory protein; SCC (cholesterol) side-chain cleavage enzyme; 3β-HSD 3β-hydroxysteroid dehydrogenase; 17β-HSD 17β-hydroxysteroid dehydrogenase
Prevalence
Most enzyme deficiencies that cause CAH are extremely rare, except for 21-hydroxylase deficiency and 11-hydroxylase deficiency. The prevalence of classic 21-hydroxylase deficiency worldwide is 1:10.000 to 1:18.000, as derived from results of neonatal screening. Non-classic 21-hydroxylase deficiency is estimated to be more prevalent (1:600), but this diagnosis can easily be missed in males because signs of adrenal androgen excess may go unrecognized. The estimated prevalence of 11-hydroxylase deficiency is 1:100.000 [1].
Presentation and Diagnosis
CAH represents a broad spectrum of phenotypes depending on the severity of the enzymatic defect. In clinical practice, however, CAH is divided into 3 forms. The most severe, classic CAH phenotype is subdivided into a salt-wasting form (SW-CAH, no residual enzymatic activity) and a simple virilizing form without aldosterone deficiency (SV-CAH, residual enzymatic activity of 1–2%). Non-classic CAH (NC-CAH) has a less severe phenotype with a residual enzymatic activity of 30–50%. Patients with the mildest forms present with symptoms are caused by androgen excess only: premature adrenarche, hirsutism, menstrual irregularities, and infertility, most of which are limited to women (non-classic form) [1].
Genetics
All forms of CAH are characterized by an autosomal recessive inheritance. The genes that encode the various enzymes have all been elucidated [1]. 21-hydroxylase is encoded by the CYP21 gene. CYP21 and the homologous pseudogene CYP21P are located in the HLA major histocompatibility complex on chromosome 6, a region with a high frequency of genomic recombination. CYP21 and CYP21P are 98% identical, which explains the high mutation rate of the CYP21 gene. One-third of the described mutations are deletions and large gene conversions that were caused by recombination between the two homologous loci. The other mutations are small defects that are transferred from the CYP21P pseudogene to the active CYP21 gene, resulting from non-homologous pairing. The genotype–phenotype correlation is reported to be 80–90%, with the best predictions in the most severe and the mildest deficiencies [1, 4].
Of all types of congenital adrenal hyperplasia, 21-hydroxylase deficiency is the most frequent and the most extensively studied. Therefore, this chapter on male hypogonadism in CAH will focus predominantly on 21-hydroxylase deficiency.
Fertility in Males with 21-Hydroxylase Deficiency
Reported Fertility in Males with 21-Hydroxylase Deficiency
Reports on child rate in CAH males show a reduced fertility. Jääskeläinen et al. [5] found a child rate of 0.07 in the complete Finnish male CAH population, compared to 0.34 in age-matched Finnish males. Other authors reported parenthood only as additional information in smaller and selected patient populations. Urban et al. [6] found apparently normal fertility in men with CAH (some even untreated): They described 14 fathers among 20 male patients (aged 18–37 years), and Cabrera et al. [7] reported paternity in 2 of 30 patients (aged 17–43 years). Three more recent reports in large patient cohorts show paternity in 41, 67, and 51%, respectively, in patients who had sought fertility [8–10].
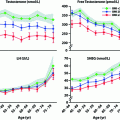
Several recent studies reported results of semen analysis in CAH patients [8, 10–13]. In most of these recent studies, only a subgroup of the study population consented to provide semen for analysis (32–91% of study participants). The combined results of these studies show azoospermia in 5–20% of cases and oligozoospermia in 20–54% of cases. In the studies in which the upper reference limit of the WHO consensus (>15 million sperm per ml) was used [14], normospermia was found between 34 and 68% [10, 12, 13]. Although reports of semen analysis are hampered by low participation rates and ambiguous use of reference values, we could conclude that spermatogenesis is impaired in a substantial group of CAH males.
In summary, there is substantial evidence that fertility can be impaired in male CAH patients. This finding raises the question to what extent male subfertility in the general population is caused by undiagnosed CAH. Ojeifo et al. [15] investigated basal and ACTH-stimulated serum 17-hydroxyprogesterone levels in a population of 50 males with idiopathic infertility, and compared the results with those of 25 controls. No differences in basal or stimulated levels were found, and they concluded that 21-hydroxylase deficiency is a rare cause of idiopathic male infertility. Pinkas et al. [16] studied a cohort of 484 healthy men attending a fertility clinic and likewise found no patient with CAH (defined as an ACTH-stimulated level of 17-hydroxyprogesterone (17OHP) of ≥45 nmol/l, performed in patients whose random mid-morning level of 17OHP was ≥6 nmol/l).
Causes of Subfertility in CAH Males
The most frequently reported cause of subfertility in CAH males is the presence of adrenal rest tumors in the testes [7, 17, 18]. These tumors may interfere directly (mechanically or paracrine) or indirectly (endocrine) with testicular function [19, 20]. Subfertility can also be caused by hypogonadotropic or hypergonadotropic hypogonadism. Psychological problems may also be important. The next sections will focus on these topics separately, although, in practice, these factors may not be separated easily.
Testicular Adrenal Rest Tumors (TART)
Introduction
The presence of testicular tumors in male patients with CAH due to 21-hydroxylase deficiency was first described in 1940 by Wilkins et al. [21]. Since then, these tumors have been described in several case reports and patient cohort studies. It is now generally accepted that testicular adrenal rest tumors are the most important cause of infertility leading to gonadal dysfunction and primary gonadal failure [22]. Because of their histological and functional resemblance to adrenocortical tissue, they are generally called testicular adrenal rest tumors (TART). These lesions are often found in both testes of adult men with CAH with a typical location within the rete testis. The tumors are benign, although case reports also describe malignant Leydig cell tumors in CAH patients that can be difficult to diagnose.
Due to the central location within the rete testis, the tumors may lead to the obstruction of the seminiferous tubules and consequently infertility. Long-standing chronic obstruction may also lead to the damage of testicular tissue, and thereby impaired Sertoli and Leydig cell function.
Prevalence of TART in Adult CAH Patients
The reported prevalence varies between 0 and 100% [5–7, 12, 17, 18, 23]. It strongly depends on patient selection (prepubertal, adolescent, or adult patients) and on the method of tumor detection (physical examination or imaging techniques). Because of the central location of the tumors, small lesions <2 cm can be easily missed. Therefore, additional techniques such as MRI or ultrasound are necessary to detect smaller lesions.
The first report of a larger patient cohort was by Urban et al. [6] who found no testicular tumors by physical examination in 30 adult patients. Using ultrasonography, however, adrenal rest tumors were described in several studies reporting TART in 2/14 adult patients [5], 9/18 adult patients [7], and 16 of 17 postpubertal patients [17]. In a recent paper with 50 adult patients (18–49 years), Reisch et al. found a prevalence of 61.3% in salt-wasting patients and 47.5% in SV patients [23].
Furthermore, there is a clear association between the prevalence of TART and the genotype of CAH [24]. TART is mostly described in patients with the most severe types of CAH. This may be explained by strongly elevated ACTH levels beginning in utero. The presence of TART in patients with non-classic CAH has only been described anecdotally [12].
Prevalence of TART in Children and Adolescents with CAH
Tumors have also been reported in childhood: Of the 8 patients with testicular tumors described by Avila et al. in a population of 38 patients, 7 were younger than 16 years old [18]. Shanklin et al. reviewed autopsy material of patients with CAH, and noted testicular nodules in 3 of 7 patients less than 8 weeks old, and in all 14 patients older than 14 months [25]. Claahsen et al. found general prevalence of 24% in childhood and adolescence [26]. Other authors found a similar prevalence of 21 and 18.3% [27, 28]. In a recent study, Claahsen-van der Grinten et al. found a high prevalence of TART in CAH children of nearly 100% with a clear increase in prevalence during puberty [29].
Diagnosing TART
The physical examination reveals only a small proportion of all testicular tumors in CAH patients. For the more accurate detection of smaller tumors, imaging techniques such as ultrasound and MR are required [6, 7, 17, 18, 30, 31]. A comparative study in CAH males concluded that ultrasound is the method of choice because it is as sensitive as MR, less costly, and more accessible [32]. Ultrasound features include hypoechoic lesions adjacent to the mediastinum testis, often lobulated and mostly bilateral (Fig. 7.2) [18, 31–38]. In larger tumors, we have observed hyperechoic reflections probably due to fibrotic strands. Tumor margins may be blurred on ultrasound but are always well defined on MR [32, 39]. On MR imaging, most of the masses are isointense on T1-weighted images and hypointense on T2-weighted images, just as are adrenal glands [32].
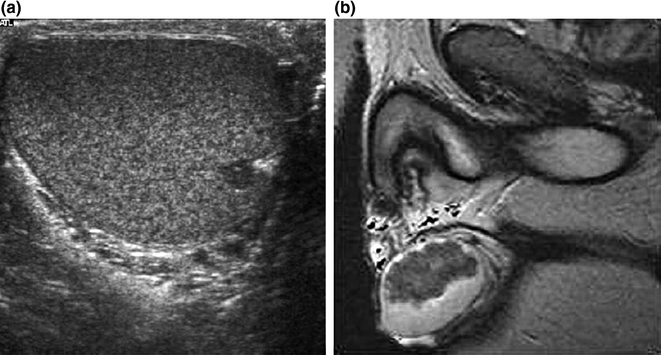
Fig. 7.2
a Scrotal ultrasound of a 13-year-old male CAH patient showing a mostly hypoechogenic rounded lesion in the left testis near the rete testis. b T2-weighted MR image of long-standing bilateral testicular adrenal rest tumor. Note that heterogeneous low-signal-intensity tumors are displaced surrounding high-signal normal testicular tissue. From Claahsen-van der Grinten et al. [22] (Open Access)
Testicular tumors in CAH may resemble malignant testicular tumors by ultrasonography and MR [38, 40]. However, clinical characteristics such as the bilateral presence of lesions and the location of the lesions adjacent to the mediastinum are helpful in differentiating between a benign testicular adrenal rest tumor and a malignant testicular lesion [33, 39, 41]. On the other hand, the ultrasound characteristics of a testicular tumor can suggest the diagnosis of CAH in males with previously undiagnosed CAH who present with a testicular mass [41]. In that case, elevated 17-hydroxyprogesterone levels may indicate 21-hydroxylase deficiency. However, 17-hydroxyprogesterone production by a Leydig cell tumor in a non-CAH patient has also been reported [42].
Histopathology
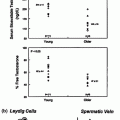
Histologically, the testicular tumors in CAH patients resemble Leydig cell tumors, and differentiation can be difficult (Figs. 7.3 and 7.4) [43]. Some differences may exist; however, TART are bilateral in 83% of cases, whereas Leydig cell tumors are bilateral in only 3% of cases. Malignant degeneration has not been reported in TART, but it occurs in 10% of Leydig cell tumors in adults [43]. In addition, TART are located in the mediastinum testis and may decrease in size when ACTH levels are suppressed by increasing the glucocorticoid dose [20, 41, 43–47]. Rich et al. [43] found that both TART and Leydig cell tumors are circumscribed, lobulated, nodular lesions. TART are typically encapsulated nodules that are dark brown (resulting from a greater concentration of lipochrome pigmentation), whereas Leydig cell tumors are usually yellow intratesticular masses. The color distinction is not always reliable, however, because some Leydig cell tumors are occasionally darker. Reinke crystals, which can be found in 25–40% of Leydig cell tumors, have never been observed in TART. In the surrounding parenchyma, seminiferous tubules show atrophic sclerosis in TART, whereas seminiferous tubules are rarely found in Leydig cell tumors because they are replaced by the tumor [47].
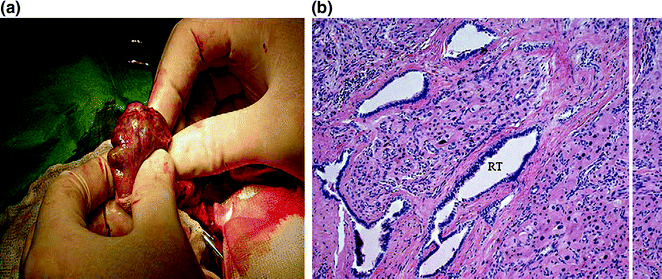
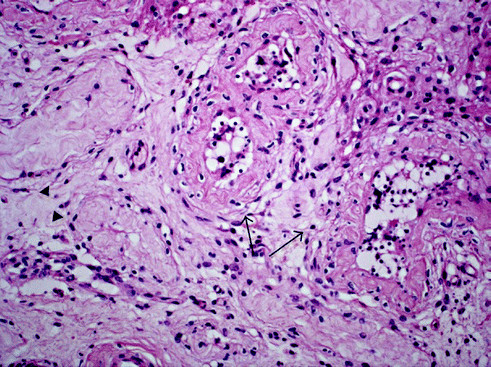
Fig. 7.3
a Macroscopic aspect of TART. Note the yellow color and the bands of fibrous tissue. b Testicular adrenal rest tumor growing into rete testis (RT) (HE, original magnification × 200). From Claahsen-van der Grinten et al. [22] (Open Access)
Fig. 7.4
Testicular biopsy of a patient showing seminiferous tubules with hypospermatogenesis and prominent peritubular fibrosis with increased number of peritubular fibroblasts (arrows), as well as tubular hyalinisation (arrowhead; original magnification × 200). From Claahsen-van der Grinten et al. [22] (Open Access)
Steroid Production
Steroid production by TART has been investigated both in vitro and in vivo.
Cortisol production by TART has been demonstrated in vivo by testicular venous sampling after stimulation with ACTH in non-classic CAH patients [48, 49] and, surprisingly, also in a salt-wasting patient [50].
17–hydroxyprogesterone production by a testicular tumor was demonstrated in vivo by testicular vein sampling after ACTH stimulation [51].
Production of 11–hydroxylated steroids, including 21-deoxycortisol (21-DF), 21-deoxycorticosterone (21-DB), and 11beta-hydroxy-Δ4-androstenedione (11beta-OHA), has been demonstrated in vitro [51, 52], as well as in vivo [51, 53, 54]. 11-hydroxylase activity is usually restricted to the adrenal cortex. Clark et al. demonstrated 11-hydroxylase activity in vitro in TART from a salt-wasting CAH patient [52]. That study identified abundant angiotensin II receptors, but no LH receptors, in tumor membranes (ACTH receptors were not investigated). In vivo, 11-hydroxylase activity was demonstrated by testicular vein sampling by Blumberg-Tick et al. [53]. Combes-Moukhovsky et al. showed that stimulation with hCG increased the secretion of 11-hydroxylated steroids [54]. LH/hCG receptors have been found in the normal human adrenal cortex [55]. In a study of 7 male CAH patients with bilateral TART undergoing testis-sparing surgery, Claahsen-van der Grinten et al. measured the concentrations of the adrenal-specific steroid 21DF, and of 17-hydroxyprogesterone (17OHP) and androstenedione (A) in blood taken from the spermatic veins during surgery [56]. In addition, mRNA expression of the adrenal-specific enzymes CYP11B1 and CYP11B2, as well as of ACTH and angiotensin II (AII) receptors, was shown by PCR in 16 testicular tumors from 8 patients [56]. The results show that TART contain adrenal-specific enzymes and produce adrenal-specific steroids suggesting that these tumors arise from adrenal-like cells. The presence at the mRNA level of ACTH and angiotensin II (AII) receptors in the adrenal rest tumors supports this hypothesis.
Testosterone production was shown in vitro by Franco-Saenz et al. [50], but only after stimulation with ACTH or hCG. Interestingly, testosterone production was more responsive to stimulation by ACTH than by hCG. The authors hypothesized the presence of specific receptors for ACTH and hCG in the same cells, a defective receptor with loss of specificity, or the presence of two different cell lines. In vivo, Kirkland et al. showed increased testosterone production after ACTH infusion into the testicular artery [57].
Benvenga et al. hypothesized from longitudinal observations in a single patient that at least three distinct types of testicular tumors in CAH could be discriminated based on their response to dexamethasone-induced ACTH suppression [58]. The first type is ACTH-dependent, both in terms of growth and steroidogenesis. The second type is only partially responsive to ACTH, since suppression of growth lagged behind the prompt suppression of steroidogenesis. In addition, this type seemed to be responsive to LH/hCG. In the third type, growth is unresponsive to dexamethasone-induced ACTH suppression, unresponsive to gonadotropins, and has lost the ability to synthesize cortisol.
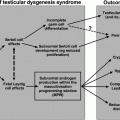
The Origin of TART
Based on histopathology, steroid-producing properties, and ACTH responsiveness, these testicular tumors are considered to consist of steroidogenic cells. It is known that cells of the adrenal cortex and gonads share a common precursor called the adreno-gonadal primordium, and develop in close relationship to each other. It was hypothesized that, due to incorrect splitting of cells, ectopic adrenal cortex cells persist and descend within the testes, and are stimulated by ACTH. Aberrant adrenal tissue has been identified in 7.5% of normal testes by Dahl et al. [59]. The aberrant adrenal cells are thought to retain their potential for glucocorticoid production, and to respond with hyperplasia to increased ACTH stimulation. Val et al. suggest that adrenal cells originate from a different cell population of adrenal-like cells migrating from the mesonephros into the developing gonad. Other studies propose that TART may be derived from pluripotent cells within the testes [60].
A more recent study by Smeets et al. [61] described the molecular characteristics of TART in more detail using qPCR of all relevant genes encoding enzymes important for steroid production. Nearly all genes were highly expressed in TART tissue, including all genes that encode the key steroidogenic enzymes. TART expression levels are generally identical to those found in adrenal tissue. The expression of adrenal cortex-specific genes (CYP21A2, CYP11B1, CYP11B2, and MC2R) in both TART and adrenal tissue is approximately 1000–10,000 times higher than in testes. In addition, the Leydig cell-specific markers INSL3 and HSD17B3 were not only found in testes, but also in TART, both at significantly higher levels than in the adrenal. The authors concluded that TART consist of a more totipotent cell with adrenal and Leydig cell-specific features [61].
Based on clinical, histological, and radiological observations, Claahsen-van der Grinten et al. [22] proposed that the development and growth of TART can be divided into five stages based on histological criteria of TART and the surrounding testicular parenchyma (Fig. 7.5). Stage 1: This stage can be defined as the presence of adrenal- like cells within the rete testis not detectable by scrotal ultrasound. Stage 2: In CAH patients, the adrenal rest cells may proliferate in the presence of increased concentrations of growth-promoting factors such as ACTH (and possibly AII). In this stage, the adrenal rest cells may become visible by ultrasound as hypoechogenic lesions. Stage 3: Further growth of the adrenal rest cells will compress the rete testis. Stage 4: Further hypertrophy and hyperplasia of the adrenal rest cells with progressive obstruction of the rete testis may lead to the induction of fibrosis within the tumor and focal lymphocytic infiltration. Several small tumors within the rete testis become confluent, forming a single lobulated structure separated from testicular tissue by fibrous strands. Stage 5: Chronic obstruction subsequently leads to the destruction of the surrounding testicular parenchyma causing irreversible damage [62]. Further studies are necessary to validate the proposed classification of TART.
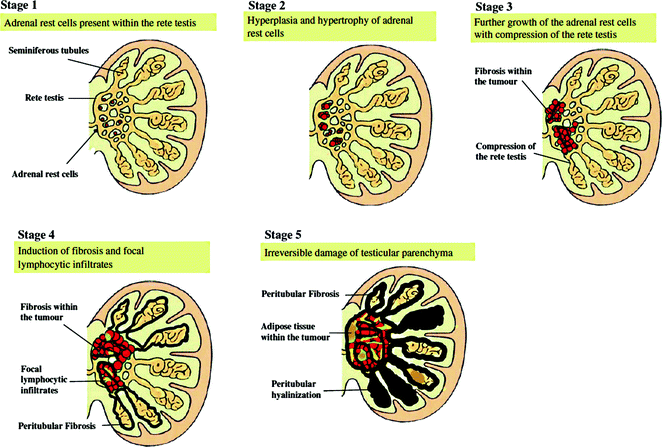
Fig. 7.5
Schematic view of the proposed classification of testicular adrenal rests. From Claahsen-van der Grinten et al. [22] (Open Access)
Relation Between the Presence of TART and Therapeutic Control
Early reports of TART were predominantly in poorly controlled patients [53, 54, 63]. Those observations led to the hypothesis that poor hormonal control and inadequate suppression of ACTH secretion is a dominant etiological factor in the development of TART. This hypothesis is supported by the development of TART in patients without CAH who have elevated ACTH levels, such as Nelson’s syndrome [64, 65] or Addison’s disease [66]. Furthermore, shrinkage of the tumor after intensifying glucocorticoïd therapy has been described in many case reports.
In more recent studies, however, no correlation was found between the presence of TART and the degree of hormonal control [7, 17, 23, 30, 31, 36]. Still, as stated before, there is a clear association between the prevalence of TART and the genotype of CAH [24]. TART is mostly described in patients with the most severe types of CAH, which may be explained by strongly elevated ACTH levels beginning in utero.
Influence of TART on Fertility
TART may interfere directly (mechanically or paracrine) or indirectly (endocrine) with testicular function. The mechanical effect of the tumors can be considerable as a result of their central location adjacent to the mediastinum testis. It can lead to the obstruction of the vascular supply, and compression and atrophy of the seminiferous tubules. Pathological examination revealed that in the cases of large tumors, the residual testicular parenchyma was abnormal and showed atrophy, sclerosis or immaturity, and decreased or absent spermatogenesis [41]. Around small nodules, however, the testicular parenchyma was generally normal [62]. Murphy et al. showed in one patient that, close to the mediastinum and the tumor, germ cells were reduced and spermatogenesis was impaired. Testicular aspiration in other areas, however, showed normal spermatogenesis. In addition to direct mechanical effects (obstruction or destruction), tumors may have a paracrine effect on the surrounding tissue, i.e., steroids produced by the tumors could be toxic to Sertoli cells or germ cells [67].
Indirect (endocrine) effects of testicular tumors in CAH on fertility could result from steroid secretion by the tumor with suppression of the hypothalamic–pituitary–gonadal axis. The association between infertility, testicular adrenal rest tumors, and depressed serum LH and FSH levels in CAH males has been described [7, 17]. However, the effect of steroid secretion by the tumor cannot be readily separated from the effect of excess steroids secreted by the adrenal glands.
Other Testicular Tumors in CAH Males
There are a few reports of other types of testicular tumors in CAH males. Adesokan et al. presented a case of a testicular adrenal rest tumor in a patient with CAH in combination with a testicular myelolipoma (a tumor of adrenal origin) and a seminoma in a cryptorchid testis [68]. Davis described a Leydig cell tumor with metastases in a CAH patient [69].
Hypogonadotropic Hypogonadism in CAH Males
In male patients with 21-hydroxylase deficiency, as in affected females, adrenal androgens may suppress the hypothalamic–pituitary–gonadal axis both directly and after conversion to estrogens, and thereby lead to hypogonadotropic hypogonadism [70–72].
Augarten et al. described a patient who was referred for secondary infertility and was diagnosed with non-classic 21-hydroxylase deficiency, as was his daughter [73]. Serum FSH and LH levels were decreased and unresponsive to GnRH stimulation. Treatment with prednisone resulted in a decrease in the serum levels of 17-hydroxyprogesterone and androgens, and normalized the response to GnRH. After 4 months of treatment, successful conception took place [73].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


