Hypoglycemia in the toddler and child
PHYSIOLOGIC DEVELOPMENT OF GLUCOSE HOMEOSTASIS DURING INFANCY AND CHILDHOOD
SYMPTOMS, SIGNS, AND EFFECTS OF HYPOGLYCEMIA
MAJOR CAUSES OF HYPOGLYCEMIA IN THE INFANT, CHILD, AND YOUNG ADULT
Genetic Disorders of Gluconeogenesis and Fasting Metabolism
Hypoglycemia in Fasting, Starvation, Illness, and Stress
Hypoglycemia Induced by Exogenous Agents
Reactive Hypoglycemia and “Spells”
FASTING SYSTEM APPROACH TO DIAGNOSIS
Introduction
Different definitions of hypoglycemia serve different purposes,1,2 and the effects of specific plasma glucose levels may vary among patients, especially those with previous hypo- and hyperglycemia. As there is no exact correspondence between risk of harm and either severity of symptoms or specific plasma glucose level, hypoglycemia should be treated as an emergency. Defensive hormonal and autonomic responses are triggered at glucose levels between 55 mg/dL and 68 mg/dL,3–6 and major neuroglycopenic manifestations can occur below 50 mg/dL.7,8 Thus a plasma glucose level of 55 serves as a reasonable threshold for diagnostic investigation and therapeutic reversal. A plasma glucose concentration between 70 and 100 mg/dL should be the target range for acute and ongoing management of hypoglycemia.
Physiologic development of glucose metabolism during infancy and childhood
Glucose utilization and production
As a meal is digested, glucose is in ample supply, but during postabsorptive fasting, glucose production must match glucose utilization rates that are markedly higher per kilogram of body weight in infants than in adults, due to their larger brain size relative to body weight.9 Bier and coworkers showed by stable isotope measurements of glucose turnover rates that the brains of infants and children use glucose at rates of 4 to 6 mg/kg/min, equivalent to almost all endogenous glucose production during fasting.10 (Figure 21-1) The rate of glucose production by the liver is linearly correlated with estimated brain weight at all ages. Muscle amino acids are the principal source of gluconeogenic precursors during fasting, but the substantially smaller muscle mass of infants relative to body mass limits the duration of fasting.11
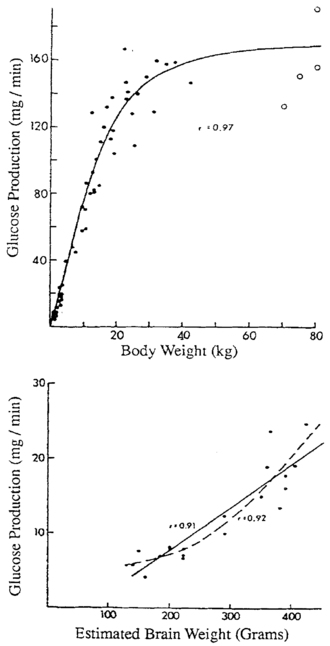
FIGURE 21-1  Glucose production as a function of body weight (top) and estimated brain weight (bottom). Note the change in slope at approximately 40 kg of body weight when brain growth is complete. (From Bier, D. M., Leake, R. D., Haymond, M. W., et al. (1977). Measurement of “true” glucose production rates in infancy and childhood, with 6,6-dideuteroglucose. Diabetes, 20, 1016.)
Glucose production as a function of body weight (top) and estimated brain weight (bottom). Note the change in slope at approximately 40 kg of body weight when brain growth is complete. (From Bier, D. M., Leake, R. D., Haymond, M. W., et al. (1977). Measurement of “true” glucose production rates in infancy and childhood, with 6,6-dideuteroglucose. Diabetes, 20, 1016.)
Because brain growth is nearly complete as the body reaches 40 kg at 10 to 12 years of age, little additional glucose production is needed in adolescents and adults, and glucose can be maintained above 70 mg/dL for progressively longer durations. Infants from 1 week to 1 year of age should be able to tolerate 15 to 18 hours of fasting before plasma glucose concentrations fall below 70.12,13 By 1 year of age, a normal child should be able to fast up to 24 hours.14–16 By 5 years of age, a fast of up to 36 hours may be tolerated, whereas most adults can maintain fasting glucose above 70 mg/dL for 48 to 72 hours.17 Hypoglycemia induced by fasting of shorter duration than expected for age should, therefore, alert the clinician to the possibility of an underlying disorder.18
Fasting adaptation to longer feeding intervals
With feeding, plasma insulin concentrations rise from values of 3 to 10 μU/mL to peaks of 20 to 50 μU/mL and stimulate glycogen synthesis, inhibit gluconeogenesis, and enhance peripheral (muscle) glucose uptake (Table 21-1). Simultaneously, triglyceride synthesis is activated and lipolysis and ketogenesis are curtailed. In the postabsorptive state, plasma glucose levels decline, and at an average threshold of 81 mg/dL (4.5 mmol/L) insulin secretion is reduced.7 Coupled with a rise in counter-regulatory hormones (glucagon and epinephrine) that begins to occur as the glucose reaches 68 mg/dL (3.8 mmol/L) and sympathetic activation at 55 (3 mmol/L), the fall in insulin levels reverses the anabolic pathways to ensure adequate supplies of glucose, fatty acids, and ketones (Box 21-1).19,20 The fatty acids mobilized from adipose tissue serve as alternative fuels for muscle, including cardiac muscle, thereby sparing glucose for brain metabolism. Glucose utilization is further spared by partial oxidation of fatty acids to ketones in the liver, which are then released to serve as substrate for the brain.21
The first phase in the metabolic defense against hypoglycemia is hepatic glycogenolysis (Figure 21-2). In infants, liver glycogen stores may provide glucose for up to 4 hours. As the child grows, glycogen reserves relative to brain glucose utilization are greater and may provide glucose for up to 8 hours of fasting. Glucagon and epinephrine, as insulin levels are suppressed, trigger glycogenolysis. Deficiency of these two hormones is unusual except in children on beta-blocker drugs. Therefore, hypoglycemia occurring early in fasting suggests either excess insulin secretion or a primary disorder in glycogenolysis.
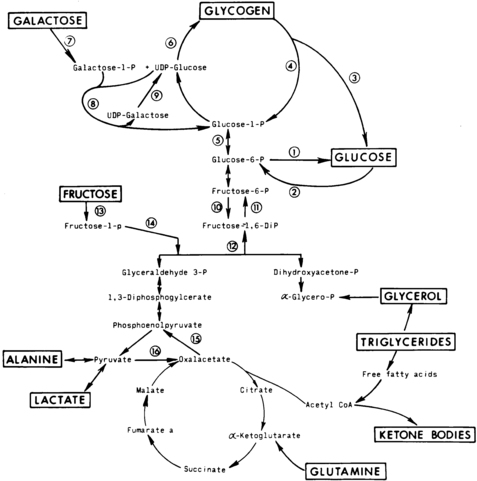
FIGURE 21-2  Contribution of major fasting systems to brain metabolism over time. Key metabolic pathways of intermediary metabolism. Disruption of the elements of these pathways may be pathogenetic in the development of hypoglycemia. Not shown is the hormonal control of these pathways. Indicated are (1) glucose 6-phosphatase, (2) glucokinase, (3) amylo-1,6-glucosidase, (4) phosphorylase, (5) phosphoglucomutase, (6) glycogen synthetase, (7) galactokinase, (8) galactose 1-phosphate uridyl transferase, (9) uridine diphosphogalactose-4-epimerase, (10) phosphofructokinase, (11) fructose 1,6- diphosphatase, (12) fructose 1,6-diphosphate aldolase, (13) fructokinase, (14) fructose 1-phosphate aldolase, (15) phosphoenolpyruvate carboxykinase, and (16) pyruvate carboxylase. UDP, uridine diphosphate. (From Pagliara, A. S., Karl, I. E., Haymond, M., & Kipnis, D. M. (1973). Hypoglycemia in infancy and childhood. J Pediatr, 82, 365–379, 558–577.)
Contribution of major fasting systems to brain metabolism over time. Key metabolic pathways of intermediary metabolism. Disruption of the elements of these pathways may be pathogenetic in the development of hypoglycemia. Not shown is the hormonal control of these pathways. Indicated are (1) glucose 6-phosphatase, (2) glucokinase, (3) amylo-1,6-glucosidase, (4) phosphorylase, (5) phosphoglucomutase, (6) glycogen synthetase, (7) galactokinase, (8) galactose 1-phosphate uridyl transferase, (9) uridine diphosphogalactose-4-epimerase, (10) phosphofructokinase, (11) fructose 1,6- diphosphatase, (12) fructose 1,6-diphosphate aldolase, (13) fructokinase, (14) fructose 1-phosphate aldolase, (15) phosphoenolpyruvate carboxykinase, and (16) pyruvate carboxylase. UDP, uridine diphosphate. (From Pagliara, A. S., Karl, I. E., Haymond, M., & Kipnis, D. M. (1973). Hypoglycemia in infancy and childhood. J Pediatr, 82, 365–379, 558–577.)
As glycogen stores become depleted, there is a greater reliance on gluconeogenesis to maintain plasma glucose levels. The main gluconeogenic precursors are amino acids, especially alanine, most of which is generated from skeletal muscle. To prevent excessive breakdown of muscle protein, adipose tissue provides an additional fuel source in the form of triglycerides hydrolyzed to glycerol, which the liver can use for gluconeogenesis, and free fatty acids, which become the major fuel source for the body at later stages of fasting. Mitochondrial fatty acid oxidation (FAO) in the liver produces ketone bodies (β-hydroxybutyrate and acetoacetate) that can be used especially by the brain but also by muscle, including cardiac muscle, for energy production (Figure 21-3). This decreases glucose utilization by these organs and helps ensure an adequate supply of glucose to the brain and to tissues that use only glucose as fuel (e.g., red blood cells). Breakdown of adipose tissue triglycerides (lipolysis) is triggered by secretion of the counter-regulatory hormones (epinephrine and growth hormone [GH]) and by declining levels of insulin. In infants, elevation of plasma ketones begins by 12 to 18 hours into fasting. In older children, ketonemia may not appear until 18 to 24 hours of fasting.22 Cortisol, produced during stress, can further accelerate gluconeogenesis.
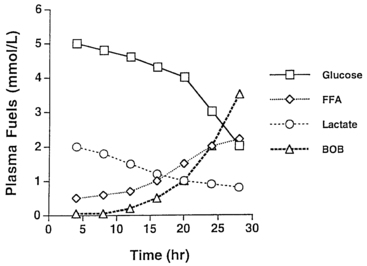
FIGURE 21-3  Changes in major metabolic fuels during fasting in a normal infant. Note that plasma glucose declines toward hypoglycemic values by 24 hours as hepatic glycogen reserves are depleted. The level of lactate, a representative gluconeogenic substrate, declines gradually during the fast. Late in fasting, levels of plasma free fatty acids (FFA) increase as lipolysis is activated—followed by an increase in β-hydroxybutyrate as rates of hepatic fatty acid oxidation and ketogenesis increase.
Changes in major metabolic fuels during fasting in a normal infant. Note that plasma glucose declines toward hypoglycemic values by 24 hours as hepatic glycogen reserves are depleted. The level of lactate, a representative gluconeogenic substrate, declines gradually during the fast. Late in fasting, levels of plasma free fatty acids (FFA) increase as lipolysis is activated—followed by an increase in β-hydroxybutyrate as rates of hepatic fatty acid oxidation and ketogenesis increase.
Symptoms, signs, and effects of hypoglycemia
Neuroendocrine defenses against hypoglycemia consist of the counter-regulatory hormones that shift metabolic processes toward glucose production and the autonomic responses that provide most of the recognizable symptoms. Counter-regulatory hormones rise as the plasma glucose drops below an average of 68 mg/dL, though the rise of these hormones is not usually clinically detectable nor rapidly measurable (Table 21-2).5,19,20,23 Of the counter-regulatory hormones, glucagon and epinephrine exert the largest immediate effects on glucose metabolism cortisol and growth hormone effects are slower, and the many other hormones (e.g., prolactin, vasopressin) that rise in response to hypoglycemia are less important still. Control of the counter-regulatory responses is at least partially local, with intra-islet control of glucagon release by release of insulin suppression, and multiple effects on glucose metabolism and eating behavior are influenced by glucose sensing in the portal vein.24–26 However, the primary site of glucose sensing and response is in the ventromedial nucleus of the hypothalamus.27
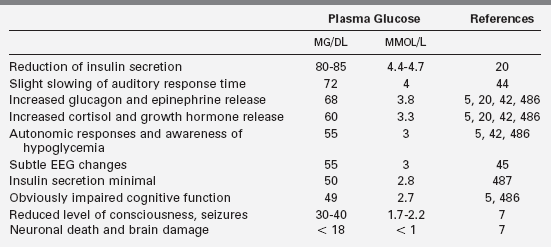
Clinically recognizable signs and symptoms of hypoglycemia occur at slightly lower plasma glucose levels and most fall into one of two categories: autonomic or neuroglycopenic (see Box 21-1). Autonomic manifestations resulting from activation of the sympathetic nervous system, typically at a plasma glucose threshold of 55 mg/dL (3 mmo/L), provide the most recognizable signal to a person that glucose is falling and food is needed.7,28 Adrenergic symptoms such as tachycardia, tremor, and anxiety are produced both by local sympathetic neural effects and by the peripheral effects of epinephrine released from the adrenals. Cholinergic symptoms include sweating, hunger, and paresthesias.28 Hypothermia often occurs with prolonged hypoglycemia in older children and adults; evidence suggests a neurogenic mechanism.29,30
The trigger for counter-regulatory responses is the plasma glucose level itself. The rate of glucose fall and the level of insulin have little effect.5,31 Activation thresholds for these neuroendocrine responses (both the counter-regulatory hormones and sympathetic activation) have been best established for healthy young adults and vary only slightly by sex,32 age,33 exercise,34 sleep,35 and nutritional status (Figure 21-4).36 Certain drugs can dampen (e.g., beta-blockers) or amplify (e.g., caffeine) the responses.37
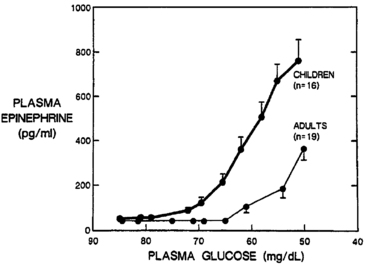
FIGURE 21-4  Plasma epinephrine response of children and adults to step decreases in blood glucose concentration during a hyperinsulinemic clamp procedure. Note that the increase in epinephrine concentration occurs at a significantly higher glucose concentration in children than in adults. In addition, at comparable glycemia of less than 60 mg/dL, epinephrine responses in children are approximately threefold higher than in adults. (From Jones, T. W., Borg, W. P., Boulware, S. D., et al. (1995). Enhanced adrenomedullary response and increased susceptibility to neuroglycopenia: mechanisms underlying the adverse effects of sugar ingestion in healthy children. J Pediatr, 126, 171.)
Plasma epinephrine response of children and adults to step decreases in blood glucose concentration during a hyperinsulinemic clamp procedure. Note that the increase in epinephrine concentration occurs at a significantly higher glucose concentration in children than in adults. In addition, at comparable glycemia of less than 60 mg/dL, epinephrine responses in children are approximately threefold higher than in adults. (From Jones, T. W., Borg, W. P., Boulware, S. D., et al. (1995). Enhanced adrenomedullary response and increased susceptibility to neuroglycopenia: mechanisms underlying the adverse effects of sugar ingestion in healthy children. J Pediatr, 126, 171.)
The most clinically important alterations of neuroendocrine response thresholds result from prior hypo- and hyperglycemia. Even a single episode of moderately severe hypoglycemia can blunt or lower the activation thresholds for 24 hours or more.38–40 Prolonged or recurrent hypoglycemia can so reduce the autonomic responses (termed hypoglycemia-associated autonomic failure [HAAF]) that neuroglycopenic effects may be the sole clinical manifestation of severe hypoglycemia.41 The effect can be demonstrated not just in adults and children with diabetes but also in nondiabetic adults and in infants with recurrent hypoglycemia due to hyperinsulinism.40 Conversely, chronic hyperglycemia is associated with higher glucose thresholds for counter-regulatory responses.42,43
Neuroglycopenic effects become progressively more severe as glucose deprivation affects brain function. For the affected person, neuroglycopenic effects are less recognizable than the autonomic symptoms and are better characterized as a continuum of increasing impairment than a single “threshold.” However, prior hypoglycemia does not lower the threshold levels for the objectively measurable neuroglycopenic effects.20 A plasma glucose decline from 87 to 72 mg/dL increases the latency of P300 waves, an auditory event-related potential said to be a sensitive and specific test of cognitive function that occurs at glucose levels above the neurogenic symptom threshold.44 Subclinical epileptiform electroencephalographic changes occur in most children at glucose levels approaching 55 mg/dL (3.1 mmol/L).45 More obvious cognitive effects, such as slower response times and impaired judgment, occur at an average threshold of 49 mg/dL (2.7 mmol/L).7,46,47 At even mildly lower plasma glucose levels, lethargy or confusion become evident, followed by seizures and coma. In infants and young children, neuroglycopenic manifestations may involve twitching, poor feeding, irritability, high-pitched crying, or even vomiting.48 With prolonged hypoglycemia in older children and adults, bizarre, uncharacteristic, or “automatic” behavior (including physical aggression or, rarely, criminal offenses) can occur.49–51 All of these cognitive, behavioral, and consciousness effects are typically completely reversed when the glucose level is raised, though subtle neuropsychological impairment may be measurable days later.52
More severe and prolonged glucose deprivation produces brain damage from neuronal death.7,53,54 In experiments with primates, 5 to 6 hours of glucose levels below 20 mg/dL (1.1 mmol/L) reliably produced severe damage.7 Severe hypoglycemia causes characteristic pathologic changes in cortical tissue and white matter, though the cerebellum and brainstem are usually spared.55 Magnetic resonance imaging changes characteristic of hypoglycemic damage can be seen in both infants and adults.56,57 Permanent cognitive impairment is measurable in many children and adults with a history of recurrent, severe hypoglycemia.58–62
Facilitated glucose transport across the blood-brain barrier, mediated by the glucose transporter-1, is dependent on the arterial plasma glucose concentration and independent of insulin. Reduction of neurogenic symptoms with repeated hypoglycemia does not depend on increased glucose transport.63 The conditions in which ketones or lactate can be used as alternate fuels during hypoglycemia are uncertain, but the ability to replace glucose is limited.64–67 A final intracerebral defense during hypoglycemia is afforded by small stores of astrocyte glycogen that can be converted to lactate and may provide up to 20 minutes of fuel support for neuronal function.68,69
Other signs and symptoms of hypoglycemia are not as clearly related to the counter-regulatory or neurogenic defenses, or to the neuroglycopenic effects. The younger the child, the more nonspecific the manifestations may be, including cyanosis, bradycardia, apnea, and apparent respiratory distress.48,70
Definition of hypoglycemia
A clinically useful definition of hypoglycemia cannot be based simply on a single value of plasma glucose concentration.1,71 For children and adults, hypoglycemia is best defined as a plasma glucose concentration low enough to elicit defensive neuroendocrine responses or to impair brain function. Counter-regulatory hormones rise at an average threshold of 68 mg/dL, and autonomic effects and the most recognizable symptoms of hypoglycemia occur at an average threshold of 55, but, as noted previously, these responses can be blunted or the thresholds shifted by prior hypo- and hyperglycemia. Brain function is slightly affected at plasma glucose levels as high as 70, and more clearly at an average threshold of 49 but these effects may be less obvious in young children.
The Endocrine Society has developed guidelines for the evaluation and management of adult hypoglycemia, which emphasize the value of Whipple’s triad for confirming hypoglycemia in adults: (1) symptoms or signs of hypoglycemia occurring with (2) a low measured blood glucose and (3) resolving when glucose is raised.71 Although this is also a useful approach for older children, Whipple’s triad may be less applicable to infants and young children, in whom the observable effects of hypoglycemia can be less specific, who are too young to report symptoms, and for whom establishing a threshold definition of hypoglycemia has been difficult.72–75 Despite the frequency with which hypoglycemia is not accompanied by obvious symptoms in infants, evidence of harm to the brain from prolonged or recurrent hypoglycemia suggests the same clinical thresholds and treatment goals are applicable to young and old alike.
Plasma glucose values must be interpreted with an awareness of numerous physiologic, technical, and artifactual sources of variation. Because whole-blood glucose levels are 10% to 15% less than plasma concentrations, it is preferable to consistently refer to plasma glucose concentrations.76 As blood from large veins will have lower glucose values than simultaneous blood from arteries, especially postprandially, it is important of refer to arterialized venous plasma glucose concentrations as the standard.77 When blood is drawn but not immediately separated, glucose levels decline due to red cell glycolysis; this is a common cause of artifactually low glucose levels reported in metabolic panels run by commercial labs.78 Glucose meters originally designed for diabetes management are useful for screening purposes, but none of the currently available meters is sufficiently accurate for a diagnosis of hypoglycemia without laboratory corroboration.79 Any meter plasma glucose level < 60 mg/dL should be confirmed by an accurate laboratory determination of plasma glucose.80
Major causes of hypoglycemia in the infant, child, and young adult
Hyperinsulinism
Hyperinsulinism is the most common and the most severe form of hypoglycemia in infancy.81–83 The hypoglycemia of hyperinsulinism is particularly dangerous to the brain because it is associated with inadequate amounts of all brain fuels (low plasma ketones and glucose).62 Although 60% of patients with congenital hyperinsulinism present in the first week of life, milder forms continue to be a significant cause of hypoglycemia throughout infancy and later. Despite a lengthening list of known genetic causes of congenital hyperinsulinism, many infants have forms for which the underlying mechanism remains unknown.84,85
Congenital hyperinsulinism often becomes evident after the first months of life, when the feeding intervals lengthen and night feedings are omitted. Such infants may present with early morning seizures or lethargy. Many will have a history of hypoglycemia in the newborn period that was not fully appreciated or will have a history of previous seizures.86 Some of these children may have previously unexplained developmental delay. In dominant forms of hyperinsulinism, a parent or other relative may report a more subtle history of hypoglycemia.87 Defects of adenosine triphosphate (ATP)-sensitive potassium channel (KATP channel) are the most common form of congenital hyperinsulinism, but a growing number of other defects in insulin secretion are being identified. As outlined in Chapter 5, all of the genetic causes of hyperinsulinism may initially present in the older infant and child.88–90
Hypoglycemia due to hyperinsulinism after the neonatal period is likely to present as recurrent episodes of neuroglycopenic symptoms in an otherwise well child. The hypoglycemia may be mild or severe, occurring after overnight fasting or in a “reactive” pattern 2 to 3 hours after a meal. Obvious adrenergic signs (pallor, perspiration) are common but not invariably present; seizures are frequent. The diagnosis of hyperinsulinism is suggested when hypoglycemia is accompanied by the absence of hyperketonemia and injection of glucagon is followed by a large increase in plasma glucose. A critical specimen obtained during hypoglycemia that demonstrates measurable amounts of insulin, C peptide, or proinsulin (Table 21-3) can be conclusive. However, it is not always easy to demonstrate elevated insulin levels.91
TABLE 21-3
Fasting Test Results Consistent with Insulinoma
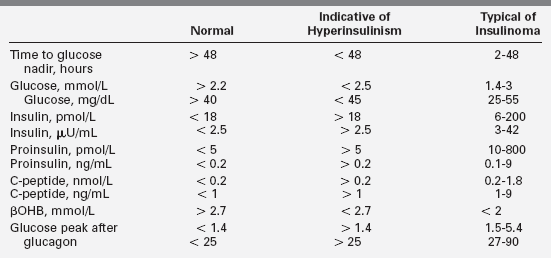
Values are compiled from several large series of mostly adult patients.193,195,198,488 Levels were obtained at time of symptomatic hypoglycemia or when glucose fell below 2.5. Levels for normal patients are at the end of a 48- or 72-hour fast. In each series, up to 3% of patients were 12 to 20 years old, though fasting longer than 24 hours is rarely necessary for the diagnosis of hypoglycemia in children. Normal ranges are somewhat assay specific and not precisely arithmetically convertible. In each series, a few patients in each category fell outside these ranges for single parameters, so diagnoses should not be based on single values near the edges of expected ranges.
Standard insulin assays may not detect exogenous insulin analogs such as lispro, aspart, glargine, glulisine, and detemir.92 Plasma free fatty acids and beta-hydroxybutyrate (βOHB) are inappropriately low during hypoglycemia due to hyperinsulinism, as is insulin-like growth-factor-binding protein-1 (IGFBP1).93 A glycemic response to glucagon at the time of hypoglycemia is strong evidence of hyperinsulinism as well.12,94,95 Because of the increased glucose utilization of hyperinsulinism, a glucose infusion rate above 8 mg/kg/min may be needed to maintain glucose above 70 mg/dL in young infants. However, this is usually more difficult to demonstrate or is inconsistently present in older children.
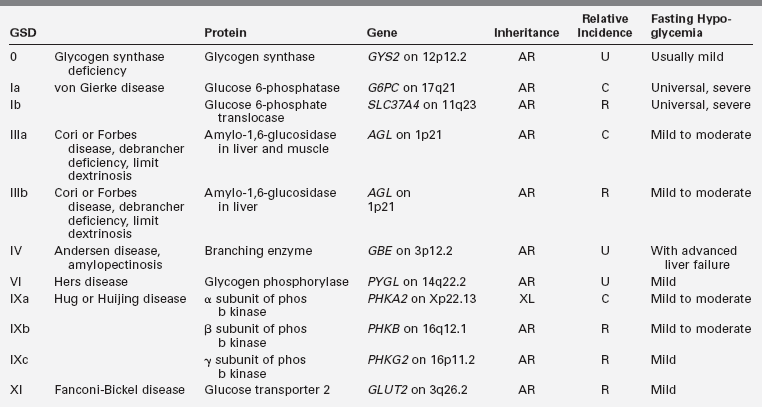
KATP channel hyperinsulinism
Two genes, SUR1 and Kir6.2, encode the two subunits of the adenosine triphosphate KATP channel in the plasma membrane of the beta cell. Potassium efflux through this channel hyperpolarizes the beta cell plasma membrane and is a key negative regulator of insulin secretion; defects that impede potassium efflux or inhibit KATP channel activity cause excessive insulin secretion. As described in Chapter 5, three distinct defects of these genes are known to cause hyperinsulinism:
• Autosomal-recessive inheritance of two KATP mutations produces a severe neonatal onset of disease that is unresponsive to diazoxide. These mutations are associated with diffuse histologic changes in the pancreatic islets.96
• A clone of beta cells possessing a single recessive mutation on the paternal chromosome may lose the normal maternal allele, resulting in homozygosity for a recessive KATP channel mutation.89 This “two-hit defect” produces a focal area of adenomatosis in the pancreas.
• Rarer, dominantly inherited autosomal mutations usually cause a milder form of KATP hyperinsulinism that often presents after infancy and responds to diazoxide. Dominant KATP mutations that produce severe, diazoxide-unresponsive disease have also been reported.97,98
The diffuse and focal forms of KATP hyperinsulinism account for more than 90% of cases of congenital hyperinsulinism in infancy and are similar in their clinical manifestations. Both may require surgery to maintain safe glucose levels. After confirmation of hyperinsulinemic hypoglycemia by the criteria presented in Table 21-3 and exclusion of the more rare forms described in the following discussion, a KATP channel defect can be presumed the most likely diagnosis. While blood is being tested for specific mutations (e.g., www.genetests.org ), management may proceed without molecular confirmation. Treatment measures able to maintain blood glucose levels above 70 mg/dL without surgery are referred to as “medical management” (Table 21-4).
Diazoxide (5 to 15 mg/kg/day) is the first-line drug, but it is often ineffective for KATP hyperinsulinism. Octreotide may be given by subcutaneous injection or infusion at 15 to 20 μg/kg/day.99 Calcium channel blockers have occasionally been tried but are generally not considered effective.100 If medical management does not maintain the plasma glucose concentration > 70 mg/dL with a normal feeding schedule, surgery may be necessary. Surgical treatment for focal KATP hyperinsulinism is curative, whereas for diffuse disease even a 95% to 99% pancreatectomy may not cure the hypoglycemia (and diabetes is a frequent sequela).95 18F-DOPA PET scanning may localize a focal lesion preoperatively, but definitive differentiation of focal and diffuse disease is made intraoperatively by repeated examination of frozen sections.94 This difficult procedure requires a patient surgeon and a dedicated team of histopathologists.101–103
Glutamate dehydrogenase hyperinsulinism
Hyperinsulinism can also be caused by activating mutations in the gene (GLUD1 on 10q) for glutamate dehydrogenase (GDH).90 This disorder, also known as the hyperinsulinism-hyperammonemia syndrome, is a milder form of hyperinsulinism than KATP hyperinsulinism and is more likely to present in late infancy and early childhood than in the newborn period.104,105 Mutations in GDH cause disease in an autosomal-dominant fashion, but up to 80% cases may be de novo mutations. Severity may vary within a family from seizures in infancy to mild postprandial hypoglycemia in adults. Ingestion of protein without carbohydrate may be especially likely to depress the blood glucose.
Activating mutations of GDH in the beta cell amplify leucine-triggered production of ATP, independent of glucose levels, which then causes closure of KATP channels and insulin release. In the kidney, the same mutation increases oxidation of glutamate to α-ketoglutarate with the production of ammonia. In addition to increased renal ammonia production, the low levels of glutamate in the liver may impair the production of N-acetylglutamate, an important allosteric activator of the urea cycle. Thus, the same mutation causes excessive insulin production in the pancreas, excessive ammonia production by the kidneys, and, possibly, impaired urea synthesis in the liver.106
The diagnosis of GDH hyperinsulinism is similar to that of other forms of hyperinsulinism. In addition to the usual laboratory findings of hyperinsulinism, however, persistently elevated ammonia levels (typically 80 to 120 μmol/L) are diagnostic of this disorder.107 Most patients respond to diazoxide, and surgery is never necessary.104,108 High ammonia levels do not appear to cause problems in GDH hyperinsulinism because the hyperammonemia is not associated with elevated neuronal glutamine, the substance thought to be principally responsible for the central nervous system (CNS) toxicity evident with other forms of hyperammonemia.109
Glucokinase hyperinsulinism
Glucokinase is the enzyme that serves as the glucose sensor in the beta cells of the pancreas.110 Gain-of-function mutations can result in a lower glucose threshold for insulin secretion, leading to persistent hypoglycemia, just as loss-of-function mutations produce a higher glucose secretion threshold, causing a common form of mild monogenic diabetes (MODY2).111 At least 15 different dominantly expressed mutations have been reported.112 Enzyme activities, glucose thresholds, and clinical severity of these mutations can vary. The more severe mutations tend to be de novo rather than inherited. The in vitro enzyme kinetics of each mutation is of limited value for predicting clinical severity and course.112
In some cases, glucokinase mutations have produced neonatal hypoglycemia severe enough to require pancreatic surgery to stabilize glucose levels. Other cases have presented as hypoglycemic seizures in childhood or even reactive hypoglycemia in older relatives. Like the KATP forms of hyperinsulinism, glucokinase mutations can cause apparently transient neonatal hypoglycemia that becomes asymptomatic for many years, only to recur later in life.113,114 In some cases, older relatives with the same mutations have had milder degrees of severity.115
Glucokinase hyperinsulinism (GCK-HI) presents some challenges to diagnosis: when glucose falls below the insulin secretion threshold, free fatty acids and βOHB can rise. In the two GCK-HI patients in whom it was tested, the acute insulin response to intravenous glucose was greater than is usually seen with KATP hyperinsulinism.86 Ammonia levels are normal, and GCK-HI patients do not have protein-sensitive hypoglycemia. The response to diazoxide has been partial in most patients. Severe cases could not be controlled on diazoxide and required pancreatectomy.116
SCHAD (or HADH) hyperinsulinism
Short chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD or HADH) is an enzyme of mitochondrial fatty acid oxidation that catalyzes oxidation of straight-chain 3-hydroxyacyl-CoAs, and is involved in multipathway complexes with other enzymes in several tissues.117 Pancreatic beta cells have relatively high levels of SCHAD activity, where it is associated with, and helps negatively regulate, GDH, the enzyme of hyperammonemic hyperinsulinism.118 Homozygous mutations of the HADH gene on chromosome 4 at q22-26 lead to an absence of SCHAD protein, deficient enzyme activity, an elevation of plasma short chain acyl-carnitine metabolites, and an autosomal recessive form of congenital hyperinsulinism.119
More than 20 patients, of at least 8 kindreds and with 12 different HADH mutations, have been reported with varying degrees of hyperinsulinemic hypoglycemia.120 Studies of the mouse model of SCHAD-HI suggest that a deficiency of HADH allows amplified activity of glutamate dehydrogenase (GDH), the same enzyme involved in hyperammonemic hyperinsulinism.118 Some cases have displayed severe hypoglycemia as newborn infants, but some showed no signs of hypoglycemia until several months of age.121,122 HADH hyperinsulinism resembles other forms of hyperinsulinism, with elevated insulin levels during hypoketotic hypoglycemia. Elevated levels of 3-hydroxybutyryl-carnitine in plasma and 3-hydroxyglutarate in urine have suggested the diagnosis in some patients. Affected patients share some features of hyperammonemic hyperinsulinism, such as a marked sensitivity to protein-induced hypoglycemia, but without the hyperammonemia characteristic of GDH-HI. Unlike other fatty acid oxidation disorders, muscle and liver function have not been clinically affected. Hypoglycemia has been severe enough in some patients to cause brain damage, but treatment with diazoxide was effective in all cases so far described.120,123,124
The oldest patient with HADH hypoglycemia was originally reported in 1977 as an instance of glucagon deficiency, in part because of a brisk rise of glucose after glucagon was given. This case was cited as an example of glucagon deficiency for years, before it was recognized that a glycemic response to glucagon was a characteristic feature of hyperinsulinemic hypoglycemia.125 Reinvestigation of the proband and other family members discovered the HADH mutation.126,127 Although this condition is usually (maybe irreversibly) referred to as a SCHAD deficiency in the endocrine literature, this nomenclature has been criticized as misleading, as another enzyme—a member of the short chain dehydrogenase/reductase superfamily with a role in brain development and perhaps Alzheimer and Parkinson diseases—has been officially and more accurately termed short chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD) for years.128
HNF4A hyperinsulinism
Hepatocyte nuclear factor 4α is a transcription factor important for pancreatic beta cell development and insulin secretion. Heterozygous inactivating mutations of the HNF4A gene are a well-recognized cause of monogenic diabetes (MODY1). Recently it was recognized that these mutations also cause excessive insulin secretion in early life, manifested by fetal macrosomia and persistent neonatal hypoglycemia. Although the diazoxide-responsive hyperinsulinemic hypoglycemia is usually transient, it may persist for several years into childhood.129 In most cases of HNF4A, the birthweight has been above average, hypoglycemia occurred in the first days of life, and a parent has had a history of monogenic diabetes.130,131 In one case, diazoxide-responsive hyperinsulinism was recognized on the first day of life, but the infant also developed hepatomegaly and renal Fanconi syndrome in the first year, suggesting that expression of the GLUT2 transporter was impaired by the defect in HNF4A.132
HNF1A hyperinsulinism
Hepatocyte nuclear factor 1 is another transcription factor important for beta cell development. Mutations can cause a common form of monogenic diabetes known as MODY 3. Two cases of mutations causing transient, diazoxide-responsive hyperinsulinemic hypoglycemia presenting at 3 and 20 months of age have been described. In both cases, the specific mutations have been reported in older patients with MODY3 and were inherited from the fathers.132
Exercise-induced hyperinsulinemic hypoglycemia
A small number of children (infants to adolescents) have had hypoglycemic seizures or syncope after intense anaerobic exercise such as soccer or swimming.133 So far, 13 patients belonging to three families have been found with exercise-induced hyperinsulinemic hypoglycemia (EIHI).134 Clinical presentations have varied, with some having severe hypoglycemic episodes from infancy, some presenting with hypoglycemic syncope after exercise as adolescents, and some only mildly affected as adults. Lab tests in severe episodes have indicated hyperinsulinism, with low ketones and free fatty acids, and inappropriately measurable insulin.134 Most patients with EIHI maintained normal glucoses during prolonged fasting. Provocative testing with brief, intense bicycle exercise induced the normal rise in plasma lactate and pyruvate levels, but affected patients’ insulin levels rose markedly for about 10 minutes after the exercise, causing hypoglycemia within the next 45 minutes.135 Intravenous infusion of 300 mmol pyruvate over 3 minutes produced a fivefold elevation of insulin within 3 minutes in patients with EIHI and has been proposed as an alternative diagnostic test. Recurrent hypoglycemia of those more severely affected was only partially preventable with diazoxide treatment.
Excessive insulin secretion in EIHI occurs because the beta cells express abnormal amounts of the MCT1 monocarboxylate transporter due to a mutation of the gene (SLC16A1) that normally prevents expression on beta cells. Although pyruvate makes an excellent substrate for ATP production, normal beta cells do not have the MCT1 transporter and hence remain unaffected by high postexercise levels of lactate and pyruvate. EIHI is inherited as an autosomal dominant condition in three of the affected families.134,136
This mechanism seems confirmed by the report of a 16-year-old boy with an insulinoma characterized by expression of MCT1 and hyperinsulinemic hypoglycemia induced by exercise and cured by resection of the insulinoma.137
Carbohydrate-deficient glycoprotein hyperinsulinism
Congenital disorders of glycosylation (CDG) are caused by deficient glycosylation of proteins or lipids.138 Many CDGs are known and more are being identified each year. Whole exome sequencing is allowing rapid identification of responsible genes,139 and a newer gene-based nomenclature system is replacing an older categorization based on transferrin isoelectrofocusing patterns.140 Hypoglycemia due to hyperinsulinism has been reported in children with three of the earliest identified defects of N-glycosylation: CDG Ia, Ib, and Id (OMIM IDs: 601785, 602579, and 601110). The initial confirmatory test for all of these forms of CDG is abnormal isoelectric focusing of transferrin.
Most cases of CDG hyperinsulinism have been identified in the newborn period, but many have been diagnosed later in infancy or early childhood. CDG hyperinsulinism is usually accompanied by manifestations affecting other organ systems—especially the brain, liver, gut, and skeleton—but cases of all three types have been reported in which hyperinsulinemic hypoglycemia was the presenting or dominant problem.141–143 The mechanism of excessive insulin production has not been determined.
PMM2-CDG (CDG-Ia) is the most common type of CDG and involves deficient activity of phosphomannomutase 2 resulting from mutations of PMM2. Many organ systems can be variably affected.144 Most patients have severe developmental delay, cerebellar hypoplasia, hypotonia, and seizures. Protein-losing enteropathy and liver disease contribute to failure to thrive. Deficient levels of antithrombin III can cause thromboses. Dysmorphic features can be subtle or obvious, including unusual fat distribution and inverted nipples. Hyperinsulinemic hypoglycemia occurs in only a minority of patients but can be mild or severe enough to warrant pancreatectomy.141,145
MPI-CDG (CDG-Ib) involves deficient activity of phosphomannose isomerase resulting from mutations of MPI. Although the sialotransferrin pattern resembles that of PMM2-CDG, the central nervous system is spared, and hyperinsulinemic hypoglycemia is a common feature, presenting in the first days of life or later in the first year.142,146 Hepatic disease and protein-losing enteropathy are usually the dominant clinical problems.147

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


