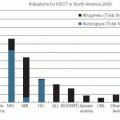
Nearly all of the available chemotherapeutic agents can produce hypersensitivity reactions (HSRs) in at least an occasional patient, and some cause reactions in 5% or more of patients receiving the drug. There are multiple agents [L-asparaginase, taxanes, procarbazine, epipodophyllotoxins, and monoclonal antibodies (MAbs)] for which HSRs are frequent enough to be a major form of treatment-limiting toxicity.
The observation and reporting of HSRs from such drugs have spurred the development of means to ameliorate or prevent this form of toxicity. For example, a chemically modified form of asparaginase (pegaspargase) was developed specifically to circumvent the HSRs and other toxicities of this cytotoxic enzyme and is marketed as a substitute for the asparaginase form derived from Escherichia coli. An example of a prevention strategy is the standard process of premedication with corticosteroids and antihistamines to decrease the incidence and severity of HSRs from paclitaxel and docetaxel.
This chapter delineates the chemotherapeutic agents that produce HSRs, the type of reaction, the clinical manifestations, and the mechanism (if identified). In addition, recommendations are provided regarding any available means of preventing or minimizing such reactions and evaluating them. The term
HSR applies to any drug-related episode with manifestations of any of the four major types of drug-induced allergy. It does not imply that all such reactions are mediated immunologically by an antibody or sensitized lymphocytes. HSRs are generally divided into four types originally defined by Gell and Coombs.
1 Table 15-1 lists these types and summarizes the mechanisms by which they are produced.
The mechanism of such reactions remains unknown for most of the chemotherapeutic agents in use. Although many HSRs from cytotoxic agents are probably immunologically mediated, there are other possible mechanisms for type I, or immediate, reactions. Some chemotherapeutic agents appear to degranulate mast cells and basophils through a direct effect on the cell surface, thereby releasing histamine and other vasoactive substances. Direct degranulation also results from the use of meperidine, morphine, codeine, curare, hydralazine, and radiopaque dyes. Some agents may activate the alternative complement pathway, also resulting in release of vasoactive substances from mast cells. Such events are not mediated by immunoglobulin E (IgE) and are termed anaphylactoid reactions. Anaphylactic reactions are by definition mediated by IgE (or sometimes IgG or IgM) and imply prior exposure to the drug or a similar antigen. Angioedema and urticaria occasionally result from stressful situations, and therefore a neural mechanism for vasoactive mediator release exists. It is not known whether any such instances have occurred in conjunction with cytotoxic drug administration. In the case of bleomycin, the rare acute reactions appear to result from direct pyrogen release from leukocytes, and no immunologic mediators are involved.
ASPARAGINASE AND PEGASPARGASE
Asparaginase is an essential element in the treatment for acute lymphoblastic leukemia, and it is the antitumor drug most likely to cause HSRs. In fact, HSRs are the principal treatment-limiting side effects. They develop in a consistent 10% to 40% of patients receiving this drug as part of combination therapeutic regimens, with the higher percentage occurring in patients who are receiving the drug either for a greater total number of doses or anew after previous therapy.
2,3,4,5,6 Rates of up to 75% for
allergic reactions have been reported without specific mention of either the reactions or their severity.
7 This high rate of HSRs is undoubtedly related to the fact that asparaginase is a polypeptide of bacterial origin that can stimulate production of IgE or other antibodies. These Igs can then mediate acute anaphylaxis with all the classic features of a type I reaction (
Table 15-1).
Evans et al.
2 studied 36 patients with asparaginase reactions. Sixteen (44%) patients had severe reactions that included respiratory distress and/or hypotension with at least a 20-mm Hg decrease in systolic BP. The other patients also had type I reactions but without hypotension. Shock and loss of consciousness, and occasionally death, have also occurred from asparaginase HSRs. The manifestations of most HSRs from this drug are sufficiently severe to warrant stopping therapy with the
E. coli-derived product when they occur.
6A number of factors enhance the risk of asparaginase-induced HSRs (
Table 15-2). The mean number of drug doses before an HSR occurred in one study was 7 (range of 1 to 11).
6 Most of the reactions will occur within the first 11 doses of the drug.
8 Immunogenicity with this drug can last at least several years after the initial drug therapy and precipitate another, and severe, reaction on reexposure.
Asparaginase is rarely used as a single agent and is most often administered concurrently with or sequentially to vincristine and prednisone and other agents. Use in such combination regimens seems to reduce the frequency of reactions in comparison to single-agent administration. Intramuscular (IM) administration also reduces the frequency of anaphylaxis,
9 but
milder degrees of type I reactions can still occur up to several hours after treatment.
10 IM administration of this drug reduces the frequency and severity of HSRs (or at least delays their onset), probably because of the fact that the pharmacokinetics of the IM route are different from the intravenous (IV) route. Maximal drug absorption takes place 10 to 24 hours after IM administration, and peak plasma levels are lower than with the IV route.
11The mechanism of asparaginase-induced HSRs is not fully understood. Khan and Hill
12 provided good evidence that IgE mediates the reaction in at least some cases. In five patients who experienced an HSR, the reactivity was mediated by a skinsensitizing IgE, as demonstrated by skin testing and Prausnitz-Kustner reaction studies.
12 Fabry et al.
13 showed that patients sustaining HSRs had evidence of complement activation with elevated levels of the C3 split product, C3d. These investigators suggested that IgG or IgM immune complexes can initiate the complement activation process and thereby mediate the reactivity.
Despite various attempts to detect reactivity before a dose of asparaginase is administered, there is no reliable way to determine which patients will sustain an HSR. Intradermal skin testing can give both false-negative and false-positive results. Test doses of small amounts of drug are also valueless. In one study,
2 an IV test dose rarely produced a reaction and was falsely negative in most patients who developed an HSR. Therefore, one must approach each dose of asparaginase to be administered as the one that could incite a serious HSR and be prepared to quickly reverse any reaction with appropriate medication. This
caveat means that antianaphylaxis medication should be close at hand when asparaginase is administered, and the patient should be observed for reactions during the hour after each dose injection. IM drug administration is preferable, when feasible, to minimize the risk of anaphylaxis.
When an HSR to the E. coli-derived asparaginase occurs, the Erwinia chrysanthemi derivative (previous species name was carotovora) is an antigenically distinct product that can be used as a substitute for continue therapy with equal antitumor efficacy. This form of the drug is not marketed in the United States, but it is commercially available in Canada and Europe. However, the U.S. Food and Drug Administration (FDA) has authorized its distribution in the United States (manufactured by HPA and distributed by EUSA Pharma) on an individual basis for those patients having an HSR to the E. coli form of the drug.
Patients who are reactive to one form of this drug have a reduced risk of being reactive to the other bacteria-derived form. However, up to 25% of patients who react to
E. coli asparaginase will cross-react to the
Erwinia derivative, particularly when multiple doses of the new drug form are
administered.
3,4,8 Precautions for HSRs must be taken when administering the substitute asparaginase, not only because of the possibility that cross-reactivity will produce another HSR from the first dose, but also that new antibodies to the substitute will develop just as readily as they did to the original form of the drug. In a few instances,
6 patients have been able to continue receiving the alternate drug form despite an HSR (usually of only mild degree) as long as appropriate premedication is administered.
Although
Erwinia-derived drug is an excellent and inexpensive substitute for the
E. coli product, patients can still suffer HSRs from it. Another (but more expensive) alternative to use when an HSR occurs is pegaspargase.
5,14 This compound is a covalent conjugate of
E. coli asparaginase with molecules of monomethoxypolyethylene glycol. To more effectively circumvent the disadvantage of HSRs and to prolong blood levels of this drug, Abuchowski et al.
15 modified asparaginase with monomethoxypolyethylene glycol at a site not involved with the antitumor activity, based on the observation that polymer conjugation to proteins could decrease their immunogenicity and extend plasma half-life. This agent showed antitumor activity in animal tumors comparable to that of native asparaginase. It is marketed for patients who develop an HSR to one or the other bacteria-derived forms of this drug. Administration of pegaspargase is more convenient for the patient because doses are given less frequently (the half-life is approximately 350 hours) and in smaller volumes. Unfortunately, patients may also develop an HSR to pegaspargase, just as they did from one or the other bacteria-derived forms, but often to a milder degree.
4,16 Data given by the manufacturer show allergic reaction rates of 10% in previously nonhypersensitive patients and 32% in previously hypersensitive patients.
17 Even when pegaspargase is used as the initial asparaginase therapy, it can still cause HSRs, but it does at a rate significantly lower than
E. coli-derived asparaginase (only 13% of patients treated in one study vs. 21% for the
E. coli product).
18
PACLITAXEL AND DOCETAXEL
Paclitaxel is now one of the most widely used drugs in clinical oncology. HSRs were a major toxicity in the initial clinical trials and an obstacle to the further development of this agent in the 1980s. Measures devised to prevent or minimize HSRs from this drug included prolonged drug infusion over 24 hours and premedication with antihistamines and corticosteroids.
19 Prolonged infusions of >3 hours have now been found to be unnecessary, and paclitaxel is even being safely administered over just 1 hour, as long as premedication is used.
20 Nevertheless, severe reactions can still occur at a rate of approximately 2%, with mild reactions (usually mild rashes or flushing) occurring in up to 40% of patients.
21,22,23The manifestations of HSRs from paclitaxel are typically type I. Weiss et al.
19 collected 27 cases of definite HSRs from paclitaxel. Forty-one percent of these patients developed hypotension, often with a diastolic pressure of zero. Rashes and dyspnea/bronchospasm occurred in approximately 75% of the patients. One patient had a fatal cardiorespiratory arrest, and such rare fatal outcomes have also been reported by others.
24 Other studies
23 have documented instances of bronchospasm sufficiently severe to require mechanical ventilation. Most (80%) patients in the series by Weiss et al.
19 had the HSR develop within 10 minutes of initiating the drug, after only a few milligrams had been infused. Only one patient sustained an HSR from a drug dose other than the first or second; one half occurred with the first treatment. In the series reported by Tyson et al.,
23 74% of the patients had the HSR associated with the first paclitaxel dose.
To prevent or ameliorate these HSRs, a three-drug prophylactic regimen (
Table 15-3) consisting of an antihistamine, a corticosteroid, and an H
2 receptor antagonist is recommended. The originally recommended means of administering this premedication
19 involved oral corticosteroids and diphenhydramine taken 12 hours and again 6 hours before paclitaxel administration. It has now been demonstrated that IV administration of such premedication half an hour before beginning a paclitaxel infusion provides an equivalent degree of protection against HSRs.
25,26,27 Prophylactic therapy does not fully prevent severe HSRs, but the reported current experience of a severe reaction rate of 2% to 3% provides good evidence it reduces the risk of such problems.
27 Weekly infusions of paclitaxel with
prophylactic therapy also have a 4% HSR rate.
28 Test doses of small amounts of the taxane are being utilized in various institutions, with varying results in limiting the severity of HSRs, being cost effective, or negatively predicting HSRs with full dose therapy. The data are encouraging enough for further study.
29,30,31During the clinical development of this drug before marketing, the recommended duration of infusion was 24 hours. However, even lengthening the infusion to 96 hours does not eliminate the risk of an HSR.
32 Shorter infusion times were clinically tested and compared in randomized manner with the 24-hour schedule.
22 Such trials have now demonstrated that the risk of an HSR is no greater with only a 3-hour infusion or even a 1-hour infusion of paclitaxel.
20 On the other hand, administering this drug in an interval of <1 hour, even with appropriate premedication, is likely to cause an inordinate risk of severe HSRs.
33The cause of these reactions has been suggested to be the drug excipient Cremophor EL, which is polyethoxylated castor oil used to maintain the solubility of paclitaxel.
19 The high frequency of hypotension in HSRs to paclitaxel and its rapid onset clinically is consistent with Cremophor effects noted in dogs, in which Cremophor and its fatty acid constituents induced histamine release and hypotension within 10 minutes of administration.
34 Cremophor EL is known to precipitate HSRs when used as the excipient with other drugs that are not antitumor agents, especially when certain techniques of drug preparation are used.
35 Szebeni et al.
36,37 have provided evidence for complement activation in vitro by Cremophor EL (in 50% alcohol as used clinically) micelles and its associated needle-like structures of crystalline paclitaxel, which provides further support to implicate this vehicle as the offending agent producing HSRs. The release of complement factors C3a and C5a can produce the symptoms of a type I HSR with no prior exposure to the drug.
This study is corroborated by the findings of another study assessing the ability of this agent to degranulate mast cells and release vasoactive substances.
38 The authors of this study
38 found only a small amount of histamine release initiated in vitro by Cremophor EL, which supports the view that mechanisms other than histamine release probably play a role in engendering HSRs from paclitaxel and its solvent. Cremophor EL-induced complement activation in serum is concentration dependent and seen in levels that are well within the range of standard dose paclitaxel.
39 The clinical features and the fact that HSRs have occurred so often with the first paclitaxel dose suggest a nonimmunologic anaphylactoid reaction—sometimes called a
C activation-related pseudoallergy.
40 On the other hand, Cremophor EL may have been falsely implicated as the etiology because docetaxel has a different excipient (polysorbate 80, also known as
Tween 80) and it produces a rate of HSRs approximately the same as paclitaxel. Polysorbate 80 has been shown to initiate histamine release both in vitro in isolated rat mast cells and in vivo in the dog with resultant hypotension,
41,42 so it too could be a contributor to reactions from docetaxel.
It appears that either the two taxanes themselves are the etiology or the drug vehicles (Cremophor EL and polysorbate 80) are equally capable of initiating HSRs. To aid in elucidating this conundrum, Essayan et al.
43 tested basophil release in vitro from leukocytes of a patient who had a documented clinical HSR from paclitaxel, comparing the cytotoxic drug alone versus Cremophor EL alone. Paclitaxel elicited histamine release, whereas Cremophor EL did not. However, similar testing by Decorti et al.
38 indicated that paclitaxel in alcohol did not stimulate histamine release, and Eschalier et al.
42 demonstrated that Cremophor EL by itself did initiate histamine release. These conflicting data still leave the issue unresolved regarding which agent (the taxane or its excipient) is responsible for causing HSRs. Perhaps the offending component is not the same in all individuals who experience a reaction. The answer is likely to be found in research
44 that is ongoing to develop alternative formulations of paclitaxel that may help lessen the risk of HSRs. The first FDA-approved alternative is the nanoparticle, albumin-bound paclitaxel, which is Cremophor EL-free. This new agent can be administered in 30 minutes with no prophylactic premedications. The total paclitaxel dose given in a course is substantially higher, with less HSRs, despite the lack of prophylactic medications.
45 These data point toward the drug vehicle as being the causative agent of a nonimmunologically based HSR.
Besides the type I reactions that are the most common manifestations of paclitaxel HSRs, other manifestations of hypersensitivity to this drug have been reported. Multiple investigators have observed cases of acute, bilateral, and transient pulmonary infiltrates developing in patients receiving treatment with paclitaxel.
46,47,48 Such problems have occurred during the infusion, within a few hours of the infusion, and up to 2 weeks after treatment. In one patient subjected to intensive investigation regarding the etiology of such reaction,
48 paclitaxel (and not Cremophor EL) was implicated as the reactant, and the mechanism seemed to be cell-mediated (type IV) delayed hypersensitivity. The release of C3a and C5a mentioned in the preceding text could also explain these infiltrates. Another manifestation of probable cell-mediated immunogenicity that has been observed from paclitaxel is a bullous fixed drug eruption, in this case localized to the thighs.
49 A case of a similar erythematous rash because of paclitaxel with pustular eruptions, but diffusely involving the trunk and extremities, has also been reported.
50Paclitaxel is an effective antitumor agent for various cancers, and the question arises whether it can be safely administered again after an HSR occurrence. The patient may have experienced a reaction from the first dose of this drug, and if it cannot be safely given again that patient may lose an opportunity to have a beneficial antitumor response. Peereboom et al.
51 evaluated the risk of further problems in eight patients who all had developed severe manifestations of HSRs from paclitaxel. The patients all had more intensive premedication dosing, and the drug infusion rate was reduced to 10% or 25% of the usual rate for the first several hours. Five of these patients had no further HSR manifestations, and the other three had only minor symptoms.
51 Others
52 have also been able to administer more paclitaxel after an HSR, if sufficient corticosteroid premedication
is administered. However, such an approach has not met with uniform success.
53 Therefore, rechallenge with paclitaxel after an HSR should be individualized based on the need to administer more drug and testing of these various methods of minimizing the sensitivity.
The paclitaxel analog docetaxel has a similar spectrum of antitumor activity with some differences in toxicity. However, one toxicity where there is similarity is HSRs. When docetaxel was first evaluated in clinical trials, it was assumed that premedication to prevent HSRs was unnecessary because the excipient was polysorbate 80, not Cremophor EL. As discussed earlier, Cremophor EL may be the offender in HSRs from paclitaxel, and because docetaxel was not formulated with Cremophor, such reactions were assumed to be unlikely. That assumption has proved incorrect because docetaxel can initiate HSRs with approximately the same frequency and same manifestations as paclitaxel.
54,55,56 Most of these reactions have occurred with the first or second dose of docetaxel, similar to those from paclitaxel. When HSRs became a treatment-limiting toxicity with this drug, premedication with corticosteroid regimens similar to those used with paclitaxel was implemented, and the frequency and severity of HSRs have been reduced, but not to zero.
57,58 In one reported case,
58 cromoglycate administration eliminated the HSR problem after failure with corticosteroid and antihistamine premedication. Ardavanis et al.
59 reported a series of 40 patients with pre- and posttreatment tryptase and histamine levels showing no major elevations in the 22.5% of patients having HSRs. This corresponds to the recent findings of nonimmunologically based, paclitaxel-associated HSRs.
Another acute toxicity from this drug is the development of erythematous or edematous skin plaques that begin on the extremities and may spread to the trunk.
60 These lesions may begin within a few days of drug administration and may be pruritic. They are probably more a manifestation of direct skin toxicity from docetaxel, rather than an HSR. However, there may be some overlap of the manifestations of this skin toxicity and a type I reaction. Treatment has been topical steroids.
60
TENIPOSIDE AND ETOPOSIDE
Teniposide and etoposide, semisynthetic derivatives of podophyllotoxin, have a similar antitumor efficacy spectrum, although teniposide is used most often for pediatric malignancies, whereas etoposide is used for cancers in adults. Teniposide has been in clinical use for 30 years, and HSRs have long been recognized as one of its toxicities. In the last 20 years, HSRs have also been reported from etoposide, but perhaps with a lesser frequency.
Teniposide causes HSRs in approximately 6% of patients overall.
61 However, the incidence of reactions in patients with neuroblastoma can be much higher,
61,62 up to 13%. Why patients with craniospinal tumors or neuroblastoma are more likely to have HSRs from teniposide is unknown. As O’Dwyer et al.
61 pointed out, it is not a drug-dose relationship because higher doses are used for treatment for lymphoid malignancies. Neuroblastomas produce excess catecholamines, and these vasoactive substances may provide a milieu in which HSRs from teniposide are more likely. However, this disease specificity may be more apparent than real because Kellie et al.
63 observed a reaction rate of 41% in a group of patients with acute leukemia treated with teniposide. Many of these reactions were grade 1 in severity and may have been ignored or not reported in other studies of this drug. In addition, unusually high cumulative teniposide doses were administered. The reactions became more common in this study as the cumulative teniposide drug increased. Therefore, the apparent disease specificity of such reactions might disappear if the total drug courses and patient monitoring were equivalent in all studies of teniposide.
Reactions may occur from any teniposide dose, including the first one. In fact, a large fraction (32%) of the patients collected by O’Dwyer et al.
61 reacted with their initial exposure to teniposide. The timing of the reaction can be either after only a few milligrams of drug have been infused or up to several hours after drug administration. HSR manifestations from teniposide have included dyspnea and wheezing, hypotension or hypertension, sweating, urticaria, pruritus, angioedema, facial flushing, and rash.
61,62 No deaths have been reported. There are no known risk factors for sustaining a reaction, and few patients have any history of allergy. It is of interest that severe hypotension (presumably because of an HSR) has occurred even when teniposide is administered through the intraperitoneal route.
64Etoposide has also been clearly demonstrated to cause HSRs,
65,66 but the incidence of this toxicity is difficult to determine because of the anecdotal nature of the reports. Such isolated instances suggest a lower risk of HSRs from etoposide compared with teniposide. This fact may be because of either underreporting or a true lower incidence. Although underreporting could account in part for the lower number of cases, HSRs do not occur at an incidence of up to 20%, as has been observed in some series of patients treated with teniposide.
67 The only study reporting comparative data regarding the incidence of HSRs from these two agents is that of Kellie et al.,
63 and reactions of all grades from etoposide were less frequent. A reasonable estimate of the overall incidence with etoposide is 1% to 3%, although in one report
68 the incidence reached a high of 51%. The clinical manifestations of these HSRs are similar to those from teniposide. Although an occasional patient develops a reaction only after many etoposide doses, most HSRs have occurred from the first or second etoposide doses.
65,66,68The mechanism of these type I reactions from the podophyllotoxins is not known, but it is unlikely to be IgE-mediated because of the frequency of reactions from the first drug exposure (42% of the cases in one collected series
66). Equally unknown is the reason for the slightly lower incidence of reactions from etoposide. Because there is only a minor difference in chemical structure between the two congeners, this difference is unlikely to be the explanation. Moreover, despite the number of reported cases of HSRs from IV etoposide, there have been no published reports of reactions from the marketed
oral formulation of etoposide. In fact, one patient
69 who had an immediate reaction to the first dose of IV etoposide tolerated subsequent oral administration of the drug without further problems. One possible reason for a difference in the two analogs is that teniposide might produce a reactive metabolite in higher concentration than etoposide. Another possible explanation is the difference in the formulation of the two drugs: Teniposide is formulated with Cremophor EL and etoposide with polysorbate 80 and benzyl alcohol. Cremophor could be the inducer of the teniposide-related reactions, as is perhaps the case with paclitaxel. However, Nolte et al.
70 attempted to implicate Cremophor by studying in vitro histamine release from both teniposide and Cremophor, and only teniposide degranulated basophils. Moreover, this effect was dose-dependent and not IgE-mediated. Finally, the most compelling evidence that the podophyllotoxin itself is the inducer of HSRs is the fact that etoposide phosphate, an ester prodrug that does not contain any excipients, can also initiate HSRs.
71,72 On the other hand, there have been two reported instances in which a patient who had an HSR from etoposide did not have a cross-reaction when treated with etoposide phosphate,
73,74 so perhaps some of the observed HSRs have been caused by the polysorbate 80 excipient and not the cytotoxic agent. If etoposide itself can precipitate reactions, it is surprising the oral formulation (containing citric acid, glycerin, and polyethylene glycol) has so far not been reported to cause HSRs.
Rechallenge with teniposide has been attempted (usually after antihistamine and corticosteroid premedication) with some success, thereby allowing further drug administration.
61 Etoposide can also be substituted for teniposide with no cross-reactivity in some cases.
62 However, some patients
62 have also reacted to the etoposide given as a substitute, and this fact gives some insight into the mechanism of reactions from these two drugs. If substitution of the congener results in cross-reactivity in some cases, the inciting agent must be the chemotherapeutic agent, not the excipient. In other cases without cross-reactivity, the reverse may be true—that is, the excipient (either polysorbate 80 or Cremophor EL) may be the offending agent. As may be the case with paclitaxel and docetaxel, an HSR may be initiated in some patients by the excipient and in others by the cytotoxic drug.
In two cases, rare type II hemolytic reactions were reported secondary to teniposide.
75,76 A drug-dependent IgG was found to mediate the reaction. Despite an incidence as high as 13% for type I HSRs because of teniposide, only this single case of a type II reaction has been published.
CISPLATIN, CARBOPLATIN, AND OXALIPLATIN
Cisplatin reactions have been reported since the initial clinical use of this drug in the early 1970s.
77 There has never been a reliable determination of the incidence of cisplatin-induced reactions, but the reported rates of this toxicity seem to be less in the last two decades than in the 1970s. For example, in a study reported
78 in 1979, the rate of HSRs was 20%. There are no reports of patient series treated more recently with anywhere near this rate. Several possible reasons may explain this change. One reason is that antiemetics are liberally prescribed as premedication for any patient receiving cisplatin, and dexamethasone and diphenhydramine are often used for this purpose, perhaps suppressing any HSR while minimizing vomiting. Another reason is that no patient in the study by Gralla et al.,
79 which had a 6% rate of reactions, developed hypersensitivity before receiving six or more doses of cisplatin. Cisplatin is usually given now for a fewer number of cycles, and immunogenic reactivity may not have time to develop. If the number of cisplatin cycles exceeds six, the risk of reaction may rise. Whatever the reason, the frequency of HSRs from cisplatin is much less than it used to be, although anecdotal reports continue to be published.
80,81IV cisplatin can cause HSRs, and so can drug instilled intravesically for bladder cancer. The incidence of these latter reactions is much higher than with IV administration. For example, in a study of intravesical cisplatin use by Denis
82 7 (10.4%) of the 67 reported patients sustained HSRs from intravesical cisplatin. The minimum number of repetitive drug instillations before a reaction occurred was eight, given over 4 months. There was an incidence of 14% among those patients who received a minimum of 4 months of treatment. These data support the hypothesis that a certain minimum amount of cisplatin courses (usually six or more) is necessary to induce reactivity. Cisplatin administered intraperitoneally may also induce HSRs, even with the first dose.
83From the above data, one could conclude that cisplatin administered intravesically (presumably involving systemic drug absorption) is more allergenic than when it is given intravenously. However, there is no logical explanation for such a difference. It may be the fact antiemetic premedication with corticosteroids would not be routinely used with this route of cisplatin administration.
The manifestations of the type I HSRs to cisplatin, whatever the route of administration, are anxiety, pruritus, cough, dyspnea, diaphoresis, angioedema, vomiting, bronchospasm, rashes and urticaria, and hypotension. Prompt administration of corticosteroids and antihistamines usually effectively aborts any reaction. Several deaths have been reported
84,85 from a cisplatin-induced HSR, but the first
84 patient may have had a very unusual platinum sensitivity. She previously had a seizure from carboplatin, so when cisplatin was to be given as a substitute she was heavily premedicated against having an HSR. She sustained one anyway despite receiving only a very small cisplatin dose.
The mechanism of cisplatin-related type I reactions has been investigated in only a few patients. Khan et al.
77 demonstrated that at least one case of cisplatin-induced reaction was mediated by IgE. In contrast, Wiesenfeld et al.
86 studied a patient who sustained a reaction for evidence of IgE-mediated reactivity and found none. Goldberg et al.
87 skin tested two patients after reactions to cisplatin, and both had skin reactivity to the drug, whereas six control patients, who also received cisplatin therapy, did not. Type IV reactions (contact skin reactions) have also been reported from occupational exposure to cisplatin.
88When analyzing a patient who sustains an HSR, apparently from an antitumor agent, one must always consider the
possibility that the drug formulation vehicle (the excipient) or other drugs administered concurrently are the allergens and not the antitumor agent. In the case of cisplatin other agents, such as antiemetics, are frequently administered with it. Historically diuretics and mannitol have been coadministered. Mannitol, by itself, may cause HSRs in rare instances.
89 One patient
90 clearly had an HSR to mannitol (given to promote osmotic diuresis) administered just before cisplatin was to be infused. Mannitolassociated anaphylaxis has been demonstrated in one patient to be related to mannitol-specific IgE.
91 In addition, ondansetron, which is used frequently as an antiemetic with cisplatin, is also capable of inducing an HSR.
92 It is possible that some of the historically reported HSRs ascribed to cisplatin have been caused by mannitol administered as an osmotic diuretic with the cisplatin.
Type II reactions with hemolytic anemia have been reported in a few patients treated with cisplatin.
93 The hemolysis was proved to be caused by cisplatin by appropriate laboratory tests (direct antiglobulin testing using polyspecific and monospecific reagents) and hemolysis recurrence with drug rechallenge. However, one must be cautious in ascribing possible hemolytic episodes to the presence of cisplatin-induced antibodies. Zeger et al.
94 have shown that cisplatin can cause a nonimmunologic binding to red cell membranes and γ globulins, thereby giving a false-positive direct antiglobulin test even in the absence of any hemolysis. Anemia is a common toxicity from cisplatin, especially if it is administered repetitively, and treatment with erythropoietin and/or transfusions may be required. However, this anemia is related to both the chronic myelotoxic effect of cisplatin and an erythropoietin deficiency from renal tubular damage by the drug. In only rare instances it is caused by hemolysis, but a sudden decrease in hematocrit when using cisplatin should raise suspicion of a type II reaction. Appropriate studies should then be performed to evaluate this possibility.
Carboplatin is a cisplatin analog with nearly equivalent antitumor efficacy and lower rates of some toxicities. Over the last decade, it has been found to be effective in a growing number of cancers and use has increased, along with reports of HSRs. Several instances of carboplatin-induced HSRs have been published.
95,96,97,98,99,100,101,102,103,104,105 More recent series
102,103,104,105 indicate that the incidence of carboplatin HSRs varies from 2% to 50%, which is decidedly higher than those reported for cisplatin The manifestations of these carboplatin reactions are typical type I, although as with cisplatin there are rare reports
106 of immune (type II reaction) hemolytic anemia being induced by carboplatin, and two cases
107,108 of coronary vasospasm associated with carboplatin infusion. McAlpine et al.
109 report 14 patients with delayed symptoms of up to 14 days after exposure. The more common findings were rash, bilateral palmar itching, and facial symptoms. Ten of the patients (83%) converted to positive in skin testing, with seven converting immediately. With few exceptions,
110 the HSRs have occurred after considerable prior exposure to carboplatin and in some cases cisplatin also. In a series reported by Markman et al.,
99 the median number of platinum cycles received before the HSR occurred was 8 (range of 6 to 21). The risk of such reactions appears to rise precipitously after six cycles of therapy. In a series reported by Morgan et al.,
95 67% of the patients receiving 10 cycles had HSRs. Others have also seen reactions only after very large cumulative doses of carboplatin.
96Patients have been reported who had a type I HSR to cisplatin and then developed a similar reaction to carboplatin,
100 and vice versa,
111 when the alternate analog was used as substitute therapy. It is clear from these cases that there is a risk of cross-reactivity from these analogs when one is substituted for the other after an HSR. In addition, some of the reported patients had been treated with cisplatin previously without any problem, and then after receiving only a few doses of carboplatin, developed a life-threatening HSR to it.
112 These patients were probably sensitized to the cisplatin, and only a few doses of carboplatin were necessary to precipitate a reaction.
The mechanism of reaction to carboplatin has been studied in detail in one case.
113 The patient had skin test reactivity to both carboplatin, which precipitated the HSR, and to cisplatin, which she had received previously. For technical reasons, the investigators
113 were unable to test for specific IgE, but the patient did have a high level of serum IgE a month after the HSR, which fell to normal 3 months later.
A major concern with both platinum derivatives is whether additional therapy can be administered despite the occurrence of an HSR. It has been both possible
96,97 and impossible
95 to substitute the analog when a reaction has occurred. It has also been possible
95 and impossible
87 to administer more of the same agent after premedication with corticosteroids and antihistamine without further HSRs. Desensitization before rechallenge has been used successfully in a few cases
98,110 when it was desired to continue therapy with the analog that had caused a reaction.
Oxaliplatin was initially used in some European countries and licensed in the United States in 2002 for use in advanced colorectal cancers. As with the other platinum agents, the main reactions are type I with respiratory and cutaneous symptoms predominating and appearing soon after the infusion begins.
114 There has been much thought about the possible mechanisms that could include IgE, polyclonal T-cell expansion acting as superantigens, oxaliplatin binding to major histocompatibility complex, or just local tissue damage from the drug.
115 Although all could contribute, only the role of IgE in the process seems to be confirmed. Meyer et al.
116 reported positive skin testing in a majority of the eight patients (6/8) tested and all were negative for cisplatin. Herrero et al.
117 also performed skin testing in five patients, all of whom were negative for their prescribed antiemetics and positive for oxaliplatin.
The initial incidence
118 of HSRs during clinical trials was 0.55%, but with routine use, the incidence is now noted
115,119 to be between 10% and 13%. There have been no reports of HSRs with the first infusion of oxaliplatin. The average number of cycles till onset of HSRs
115,120 has been between 7 and 9.4. Most cases of severe HSRs follow the typical pattern and are associated with hypotension, but there is a report of two patients who experienced a combination of unconsciousness and hypertensive crisis.
121Type II reactions with hemolytic anemia have been reported.
122,123,124,125 The hemolysis has been proved to be caused by oxaliplatin by appropriate laboratory testing. In three cases,
122,123,124 rechallange with oxaliplatin precipitated the hemolysis again, one time with fatal results.
122 A single type III reaction has been reported.
126Rechallange after HSR can be accomplished with antihistamine and steroid premedication, but it appears that most patients will reexperience symptoms.
115 Meyer et al.
116 were the first to report a successful desensitization protocol in patients with HSRs because of oxaliplatin. The protocol was based on successful trials to desensitize patients to carboplatin, using increasing dilutions and prolonged infusion times. A patient treated by Bhargava et al.
127 was able to undergo six additional cycles of therapy after her similar desensitization program. In order to prevent HSRs, antihistamine and steroid premedication has been employed with good success, but lengthening the infusion time is also of benefit. Longer infusion rates seem to prevent the onset of HSRs, as indicated by the lack of HSRs in 12-hour infusions or 5-day continuous infusions.
128 Five patients who had HSRs with 2-hour infusions did not have symptoms when given 6-hour infusions
129 and Lee et al. reported a patient who had severe HSR tolerating five subsequent courses of an oxaliplatin regimen by doubling the infusion time.
121
PROCARBAZINE
Procarbazine has been known to produce HSRs since its initial clinical use in the early 1960s. The most common manifestation of reaction is a diffuse, pruritic, erythematous maculopapular rash, but urticaria, angioedema, and high fever have also been observed.
130,131 Two reports have suggested such reactions occur with some frequency, particularly in patients being treated for brain tumors. Skin reactions have been observed in 12% to 35% of patients treated for gliomas with procarbazine.
131,132,133 Other procarbazine-related skin reactions include toxic epidermal necrolysis
134 and fixed drug eruptions.
135 Procarbazine can also produce rare instances of pulmonary toxicity with features of type III reactions.
131,136,137 The patients with this form of toxicity have interstitial pneumonitis with eosinophilia in the peripheral blood and/or lung biopsy, and they improve with procarbazine withdrawal and corticosteroid therapy.
The mechanism(s) of these HSRs from procarbazine has been poorly studied and is not known. Only two patients have been studied in detail in an attempt to define a mechanism. One patient
130 was found to have a low complement level, possibly because of depletion of complement fragments from release of anaphylotoxins or the formation of immune complexes. The other patient
135 clearly had a fixed drug skin eruption, but the pathogenesis of such reactions has not been determined. Skin patch testing with procarbazine in this patient was nonreactive. However, this sort of test is not necessarily reliable because the HSRs may be because of a procarbazine metabolite and not the parent compound. The reason patients with gliomas have a high incidence of reactivity to procarbazine may be the frequent concurrent use of anticonvulsants for such cases with some undetermined interaction between the two drugs.
138Many of the reported patients have been rechallenged with procarbazine after suspension of treatment and resolution of the symptoms,
130,135 and their HSR manifestations promptly recurred. Two patients who were rechallenged had severe reactions.
131 One patient
137 was rechallenged twice with procarbazine and had a recurrence of pneumonitis each time. It appears that procarbazine cannot be safely readministered, even with the use of concomitant steroids and antihistamines to minimize further HSRs. Patients who receive procarbazine in the mechlorethamine, vincristine, procarbazine, and prednisone regimen for Hodgkin disease are already receiving prednisone as part of the chemotherapeutic regimen. HSRs to procarbazine may still occur despite this corticosteroid therapy.
139 The only apparent means of preventing such reactions, once they develop, is to discontinue the procarbazine permanently. Whenever skin reactions to procarbazine occur in a setting where allopurinol is used concurrently, one must be certain that the allopurinol is not the initiator of the reaction because this drug is well-known to produce diffuse skin reactions similar to those related to procarbazine.
140
CYTARABINE
Although cytarabine has been in clinical use for 35 years, HSRs have only recently been observed with more than anecdotal frequency. This change may be because of the fact that the cytarabine doses now being widely used are 10 to 15 times greater than those used before the 1980s. Four distinct forms of HSRs to this drug have now been recorded.
The first form of HSR is type I, with dyspnea, chest pain, fever, maculopapular rash, urticaria, and hypotension.
141,142,143,144 The most common manifestation of reaction has been a rash, but anaphylaxis with hypotension to shock levels has occurred in a few patients.
144 The mechanism of type I reactions has been studied in three patients,
141,143,144 and evidence was presented that implied the reactions were mediated by IgE.
Another acute reaction induced by this drug has been termed the
cytarabine syndrome by Castleberry et al.
142 The manifestations of this type of reaction are high fever, rigors, diaphoresis, myalgia, arthralgia, and maculopapular rash. Other patients
145 have been reported to develop similar acute reactions, with one having hypotension reaching shock level and requiring vasopressor support.
146 This cytarabine syndrome may or may not be a form of HSR. Williams and Larson
146 suggested that this was a form of an HSR because of the presence of circulating immune complexes in their patient (perhaps a type III reaction?). However, no studies have been done to investigate this possibility. Although an immunologic basis for the manifestations of the cytarabine syndrome is possible, a more likely mechanism is direct cytarabine toxicities with no mediation by antibodies.
The frequency of this acute reaction varies from isolated case reports to as high as 33%
142 of patients treated with cytarabine. It is probably more common than the rare instance for which a case report is published. Patients with acute leukemia have leukopenia and often become septic, with associated fever,
myalgia, diaphoresis, rigors, and even hypotension. When such symptoms occur, they are probably often ascribed to sepsis, and the possibility of a cytarabine toxicity may not be appreciated.
The type I reactions described from cytarabine can be initiated by either moderate or high doses of drug. However, a third type of reaction induced by cytarabine seems to be generally dose related. First described by Burgdorf et al.,
147 this reaction is characterized by intense, painful, erythema of the palms, fingers, and soles, with later development of bullae and desquamation.
148,149 Dysesthesias often herald the onset of the erythema, and the edema may be marked enough to limit finger motion. The erythema begins a median of 6 days after initiating cytarabine therapy. Some patients also have had fever, facial edema and erythema, and/or erythematous maculopapular rashes characteristic of a type I reaction. In most cases only the hands are involved, but this exanthem has also been described as involving the eyes and lips
150 and even only the ears.
151 If this reaction occurs in the setting of high-dose cytarabine and allogeneic marrow transplant, it must be differentiated from the severe skin reaction that may be part of graft-versus-host disease.
152 Skin biopsies have been obtained from some patients at the site of the reaction.
149,150,152 The histologic abnormalities are nonspecific and are typical of drug-induced skin eruptions.
The reaction is usually self-limited, but steroids have also been used to hasten resolution.
153 The skin usually heals without scarring. Some patients have been treated with cytarabine again without recurrence of this toxicity,
149 but in other series
150 it has recurred after subsequent chemotherapy.
The incidence of this palmar-plantar reaction has varied from 6% to 40% of patients treated with cytarabine doses of 1,000 mg per m
2 or higher,
150,152 especially if eight or more doses at this level have been administered. Some patients treated with conventional cytarabine doses of 100 mg per m
2 have also developed this acral erythema
149,154 in rates up to 40%.
The mechanism of these skin reactions involving primarily the palms and soles is not known. It may be a vasculitis that peculiarly involves certain small peripheral blood vessels, possibly because of a type III reaction. However, the relationship to drug dose is more indicative of a direct cytotoxicity to skin and subcutaneous tissues. In addition, this phenomenon is not limited to patients receiving cytarabine. It has also been observed in patients receiving doxorubicin, hydroxyurea, cisplatin, cyclophosphamide, and 6-mercaptopurine.
152,155The fourth type of cytarabine-induced reaction is a dermatosis in which histologically there is a marked accumulation of neutrophils around the dermal sweat glands, termed
neutrophilic eccrine hidradenitis.
155,156 The skin lesions are erythematous plaques or nodules that may be tender and are not pruritic. The mechanism of this apparent drug-induced problem is not known. Flynn et al.
155 suggested this problem is a form of an HSR, but it is more likely a variant of the direct cytotoxic effect cytarabine may have on skin tissues.
ANTHRACYCLINES
Doxorubicin has occasionally produced severe and generalized type I reactions.
157,158,159 These HSRs are characterized by urticaria, pruritus, angioedema, dyspnea and bronchospasm, and sometimes hypotension. Such reactions have even occurred when doxorubicin was administered intravesically,
160 presumably because of systemic drug absorption. The liposomal formulation of doxorubicin can also initiate a type I reaction, even on the first dose.
161 Daunorubicin has also produced rare instances of such reactions,
162 but its close analog idarubicin has not been reported to cause an HSR from either the IV or oral formulations. Epirubicin has been reported to cause pruritus and rash even with steroid premedication, and anaphylaxis evidenced by hypotension and hypoxemia.
163,164 In the case of doxorubicin, dexrazoxane is sometimes administered with it for cardioprotective effects, and it has caused HSRs in rare instances.
165Doxorubicin can also cause erythema, pruritus, and urticaria localized at, or adjacent to, the drug injection site.
166 This phenomenon is referred to as a
flare reaction. Although it can raise concern the drug was extravasated or that more serious allergy manifestations will develop, it does not seem to progress to a generalized HSR, and it is not because of anthracycline extravasation. It is usually transient and disappears without treatment. Further drug can be given later without concern for more serious problems because the reactions often do not recur, and even if they do, they are not more intense or generalized. To obviate the concern for a worse reaction, one can administer premedication with antihistamines and corticosteroids, which will prevent further problems. Doxorubicin analogs such as epirubicin are also known
167 to produce such flare reactions.
Another form of skin reaction that can occur from liposomal doxorubicin is palmar-plantar erythrodysesthesia, also known as the
hand-foot syndrome.
168 However, this reaction is probably not an HSR but a direct dermatologic toxicity of this doxorubicin formulation, which is designed to prolong blood levels of the cytotoxic agent. Oral dexamethasone has been successfully demonstrated to alleviate or eliminate the reaction and allow therapy to continue without delay or dose
Although the cytotoxic antibiotic is undoubtedly the initiator of these HSRs, the drug manufacturer recommends reconstitution of the lyophilized doxorubicin with saline (instead of distilled water) to reduce the frequency of the localized flare reactions. However, Solimando and Wilson
157 demonstrated that systemic type I HSRs are not inhibited by the use of saline reconstitution. Chanan-Khan et al.
170 have recently investigated the HSR rate and complement activation in liposomal doxorubicin infusions. Similar to paclitaxel, they discovered that liposomal doxorubicin can activate complement and does so at a rate of 72% in the study group of 29 patients who were not given premedication. The HSR rate was 45% and correlated to the plasma levels of protein S-bound C terminal complexes that were significantly elevated in 92% of those with reactions and 56% in those without HSRs.
CYCLOPHOSPHAMIDE AND IFOSFAMIDE
Although cyclophosphamide is immunosuppressive, it can cause antibody-mediated type I reactions (including
anaphylaxis and shock) in rare instances.
171,172,173,174 In addition, cutaneous vasculitis (a type III reaction?) has been observed.
175 HSRs have occurred from both oral and IV administration of cyclophosphamide but more often the latter, and they have occurred both immediately after drug administration
171,172,173 and hours later.
174Cross-reactivity with other alkylating agents is variable. Some patients reactive to cyclophosphamide have tolerated chlorambucil as a substitute without further reactions,
173,175,176 but at least one
177 did not. Another patient was treated with ifosfamide after a reaction to cyclophosphamide and did not cross-react.
171 The alkylating agents have similar chemical structures. Why cross-reactivity does not occur in some patients is unclear, particularly in the case of the two analogs, cyclophosphamide and ifosfamide.
The mechanism of cyclophosphamide HSRs appears to be IgE-mediated, based on studies in a few patients.
172,178 Several investigators
172,174,178 have found that cyclophosphamide metabolites are the antigenic determinant rather than the parent compound. Cyclophosphamide must be activated in the liver, so the fact that a reaction develops up to 16 hours after drug administration
174 is consistent with the fact a metabolite is the antigen rather than parent drug.
The paradox of cyclophosphamide, an immunosuppressive agent, stimulating IgE production may be explained by the fact that in animals this drug can potentiate IgE synthesis.
176 Drug inhibition of T-suppressor cell activity on IgE B cells enhances the activity of the B cells and more IgE is produced. Therefore, the immunogenicity of an agent with otherwise low reactivity may be actually promoted in some patients.
Ifosfamide is a cyclophosphamide analog that has a similar spectrum of antitumor activity. It causes more chemical cystitis than cyclophosphamide, and it can only be used in conjunction with mesna, which reduces the incidence of the cystitis. Type I reactions have been reported
179,180 when ifosfamide and mesna have been used concurrently. Both drugs were given together, as they must be to minimize cystitis toxicity, so it is not possible to know which agent caused the reaction with reliability. However, there have been no reported cases of HSRs from ifosfamide alone, and mesna is known to cause both type I and type IV HSRs by itself.
181,182 Use of mesna with cyclophosphamide has also caused acute HSRs.
183,184 Rechallenge with mesna by itself indicated it as being the allergen,
184 and skin reactivity was positive for mesna in five patients so tested.
183 The mechanism of this reactivity was postulated to be an example of a fixed drug eruption, rather than an IgE-mediated one.
183