Marvin S. Reitz Jr., Robert C. Gallo
Human Immunodeficiency Viruses
Infection with human immunodeficiency virus type 1 (HIV-1) and its end stage, acquired immunodeficiency syndrome (AIDS), is the major public health challenge of modern times, with more than 25 million persons already dead and 30 to 40 million living with HIV/AIDS, most of whom are without access to therapy. AIDS was first recognized in the United States in 1981 with reports of unexplained opportunistic infections, including Pneumocystis jirovecii pneumonia and Kaposi sarcoma (KS), among homosexual men in New York and San Francisco.1–3 On the basis of the epidemiologic features, association with the loss of CD4+ lymphocytes and immunosuppression, and likely infectious cause, a new human retrovirus was postulated as a causal agent. The field of retrovirology had markedly advanced just a decade earlier with the description of reverse transcriptase (RT) and with the discovery of human T-cell lymphotropic virus types 1 and 2 (HTLV-1 and HTLV-2), the first two known human retroviruses, in 1979 and 1981 (reported in 1980 and 1982, respectively).4,5 The discovery of interleukin-2 (IL-2), or T-cell growth factor,6,7 allowed the culture of blood T lymphocytes from early cases of AIDS; by 1984, the detection, isolation, and propagation of HIV-1, the third human retrovirus,8–12 had led to the development of a diagnostic test, an increasingly detailed understanding of the molecular biology of this virus, and, most important, the introduction of a rational basis for antiviral therapy.13 After a long era of expanding research, new therapeutic combinations (RT and protease inhibitors), combined with the ability to measure circulating viral RNA and resistance to drugs, have led to a dramatically improved clinical course for persons fortunate enough to have access to therapy. More recently, inhibitors of virus–cell fusion, viral entry into the host cell, and integration of viral DNA into host chromosomal DNA are becoming available. Within a brief period, technologic advances have provided a clearer understanding of viral dynamics and the disease process, focusing attention on viral replication, host immune responses, and T-cell dynamics while confirming and elaborating the causal role of the virus. Equally dramatic has been the elucidation of how HIV enters cells, using both the CD4 molecule and a chemokine receptor as a dual-receptor system, as well as the mapping of the three-dimensional structure of the viral envelope protein. Contemporary retrovirology is largely devoted to the study of HIV-1 and of HIV-associated diseases. The molecular and cellular biology of this virus is now better understood than that of almost any other in history. Research on HIV has shown that rational antiviral therapy is possible, thereby pointing the way toward therapy for other viral diseases. Although this information has yet to be completely translated into greater progress in the areas of prevention, therapy, vaccines, and immune reconstitution for much of the developing world, where most HIV is transmitted, the Presidential Emergency Plan for AIDS Relief (PEPFAR) and other programs have greatly facilitated the institution of effective therapies in many parts of Africa.
Viruses are obligate intracellular parasites, and every aspect of the virus is in some way relevant to virus-host relationships. This chapter outlines the life cycle, molecular and cellular biology, structure, and regulation of HIV-1 and includes some discussion of pathogenesis and outcome of infection. Although the division of the chapter into sections is useful for organization of the information presented, in reality, these subjects cannot be separated from one another; from the perspective of both virus and host, the process of infection is a continuous series of connected interactions.
Origin and Classification of Human Retroviruses
Current knowledge places retroviral infection of humans as zoonoses that originated in primate-to-human species-jumping events. For HIV-1 and HIV-2, these events occurred in Central and West Africa, most likely at multiple times, with the more recent attaining major epidemic significance. Simian immunodeficiency virus of chimpanzees (SIVcpz) is the immediate precursor to HIV-1.14 It now appears likely that similar species-jumping events occurred between certain types of monkeys and chimpanzees.15
Retroviruses have been classified by a number of different biologic features into at least seven genera. Oncogenic retroviruses occur in all classes of vertebrates. The first infectious agents that produced cancer in chickens were isolated by Ellerman and Bang (1908)16 and by Peyton Rous (1910).17 These workers were considerably ahead of their time, and the biologic systems to culture and study these viruses had not yet been described. Rous eventually won a Nobel Prize in 1966 for his work. The pioneering work of Ludwig Gross in the 1950s stimulated renewed interest by demonstrating that oncogenic viruses could produce tumors in mammals,18 but for the next 3 decades, it remained orthodoxy for most scientists that human retroviruses did not exist. We now know that the pathogenic human retroviruses include lentiviruses (HIV-1 and HIV-2) and oncoviruses (HTLV-1 and HTLV-2). Human endogenous retroviruses, present in the human germ line, are often replication defective and have not yet been shown to cause any disease.
As a replication strategy, retroviruses use the reverse transcription of viral RNA into linear double-stranded DNA, with subsequent integration into the host genome. The characteristic enzyme used for this process, an RNA-dependent DNA polymerase that reverses the flow of genetic information, is known as reverse transcriptase. The discovery of this enzyme altered the “central dogma” of molecular biology—namely, that genetic information must necessarily flow from DNA to RNA19,20—and helped initiate the modern era of molecular biology. This enzyme is error prone; with the massive turnover of virions in the infected host, these errors accumulate in the viral DNA, accounting for the relatively high mutability of HIV-1. The lifestyle of the retrovirus therefore involves two forms, a DNA provirus and RNA-containing infectious virion.
The discovery of HTLV-1 and its etiologic association, first with adult T-cell leukemia, an aggressive T-cell lymphoma,21–23 and later with a neurologic disease, tropical spastic paraparesis/HTLV-1–associated myelopathy (TSP/HAM),24 were pivotal events in modern medicine (see Chapter 170). Although there is relatively little variation among HTLV-1 isolates, HTLV-2, the second human retrovirus, is 50% identical to HTLV-1 at the genomic level.4 Similarly, HIV-2, the fourth human retrovirus, was identified as a serologic variant of HIV-1, the third human retrovirus, and was isolated from patients in western Africa.25,26 Some types of SIV are so closely related to HIV-2 that they may form an overlapping continuum with recent common ancestors. HIV-2 is known to infect several monkey species, including the sooty mangabey, its natural host, and SIV has been known to be transmitted, albeit rarely, to laboratory workers. SIVs and SIV/HIV hybrids (SHIVs) have been used extensively to study animal models of immunodeficiency. Other species, including cats (feline leukemia virus [FeLV] and feline immunodeficiency virus [FIV]) and cattle (bovine leukemia virus [BLV] and bovine immunodeficiency virus [BIV]), harbor retroviruses analogous to those of humans and some African primates. HIV-related retroviruses, known as lentiretroviruses, also include the ungulate viruses, maedi-visna virus of sheep, caprine arthritis-encephalitis virus, and equine infectious anemia virus.
As RNA viruses, retroviruses have the survival advantage of great genetic diversity. As viruses with a DNA intermediate in their replication cycle, they also have the advantage of latency, as do many DNA viruses, but even more so because the DNA provirus is integrated into the chromosomal DNA of the infected cell. As a CD4+ T-cell and macrophage-tropic virus, HIV also has the advantage of reducing the effectiveness of the host immune response.
Retroviruses are typically 100 nm in diameter and contain two single strands of RNA, which permits recombination between the strands (Fig. 171-1). The typical genome is approximately 10 kb or less in size and contains three major structural genes, namely, gag (group-specific antigen), pol (polymerase), and env (envelope). HIV-1 also contains several additional genes; these “extra” genes were first described in HTLV-1. In both viruses, some of these extra genes are essential to viral replication, whereas others may modulate interactions of the virus with its host. Figure 171-2 outlines the genome composition of HIV-1 and HTLV-1.
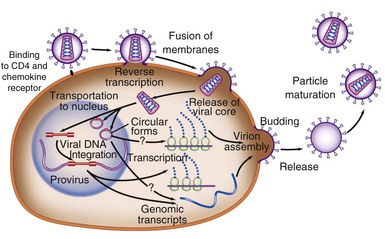

Viral Transmission and Life Cycle
Biology of Transmission
The infectious life cycle of HIV can be described both at the molecular, single-cell level and at the level of a host organism infected with a “swarm” of closely related viral species. Multiple variables, including route of exposure, dose, immunogenetic background, and concomitant infections, influence the probability of transmission. The biologic events during exposure and successful infection of the host are only partially elucidated. The most common modes of infection are sexual transmission at the genital or colonic mucosa; exposure to other infected fluids, such as blood or blood products; transmission from mother to infant; and occasionally, accidental occupational exposure. Transmitted viruses typically use the interaction of the viral glycoprotein gp120 with the cellular receptor CD4 and the chemokine receptor CCR5 to gain cell entry, thus selecting for macrophage-tropic non–syncytia-forming variants. This finding explains why persons who lack a functional CCR5 receptor are relatively resistant to infection by sexual transmission. Non–macrophage-tropic strains, also called syncytia-inducing, are typically found late in infection. These strains use another chemokine receptor, CXCR4, to facilitate entry and are apparently not readily transmitted from person to person. In a model of acute infection in the macaque, the first cellular targets of intravaginal inoculation of the virus are Langerhans cells, tissue dendritic cells in the lamina propria, which then fuse with lymphocytes. However, direct infection of T cells has also been described. Infected cells can be found in draining lymph nodes within 2 days and in plasma after 5 days.27
In HIV-1–infected persons, there is a rapid rise in the degree of plasma viremia within days, with high viral titers and widespread dissemination, probably targeting lymphoid organs and the central nervous system (CNS). This acute stage of HIV infection is sometimes manifested as a transient symptomatic illness characterized by a maculopapular rash and flulike symptoms. This phase is followed by a marked reduction in virus to steady-state levels, probably because of vigorous antivirus cellular responses. The immune response probably accounts for the mononucleosis-like acute syndrome seen in approximately half of patients. Initially, perhaps within hours of infection, at least three HIV inhibitory chemokines (notably the chemokines macrophage inflammatory protein-1α [MIP-1α], MIP-1β, and RANTES [regulated on activation, normal T-cell expressed and secreted]) may be produced. As is the case for many infections with viruses that establish chronic infections, this response is at least partially successful in controlling replication. Levels of HIV-1–specific cytotoxic T lymphocytes (CTLs) are inversely correlated with plasma viral RNA levels.28 High levels of potent CTL virus-specific cells targeted to the viral Env protein and soluble factors produced by these cells early in infection may correlate with the decline of virus, even before a neutralizing antibody can be detected.29
A great deal of variability in peak viral RNA plasma levels is seen during the first 120 days after infection. By 3 to 6 months after infection, viral levels reach a temporary steady state, sometimes called a viral setpoint. This level is highly correlated with subsequent disease progression in that lower viral steady-state level is correlated with slower disease progression. Thus, early in the course of infection, virus-host interactions are established that are predictive of subsequent disease.30
Intervention to control infection during this initial period has been shown to decrease the risk of subsequent infection in health care workers after needlestick exposure.31 Treatment of infected mothers and exposed neonates has also shown a dramatic effect in decreasing the incidence of maternal-fetal transmission. The theory underlying the use of antiretroviral prophylaxis after sexual exposure is biologically plausible but currently lacks direct proof of efficacy in controlled studies.32
Life Cycle
The life cycle of HIV-1 can be considered in two distinct phases (see Fig. 171-1). The initial early events include viral attachment, entry into the cytoplasm, reverse transcription, entry into the nucleus, and integration of the double-stranded DNA (the provirus). The second phase occurs over the lifetime of the infected cell as viral and cellular proteins regulate the production of viral proteins and new infectious virions.
Infection is initiated by the binding of the virion gp120 Env surface protein to the CD4 molecule found on some T cells, macrophages, and microglial cells. Both SIV and HIV-2 also use this molecule. CD4 was first identified as a viral receptor in a number of studies showing the susceptibility of CD4-bearing cells to infection and the ability to block infection with anti-CD4 monoclonal antibodies in culture. Transfection of human CD4− HeLa cells with CD4 DNA rendered them permissive for infection.33 Successful in vitro experiments blocking this interaction with soluble CD4 used laboratory strains adapted to cell lines and led to therapeutic attempts using immunoglobulin CD4, which were not successful. Subsequent experiments have shown that primary isolates are not sensitive to soluble CD4 and highlighted the necessity of using primary rather than laboratory-adapted isolates in studying virus-host interactions.
Early experiments demonstrated that as with other lentiretroviruses, macrophages could also be infected with HIV, but strains differed in their ability to infect T-cell lines or monocytes.34 Binding to CD4 is not sufficient for entry of HIV into human or nonhuman cells, and the fact that small changes in the V3 loop of envelope gp120 (see later) could determine tropism of the virus for either macrophage or T-cell lines suggested that a second receptor was present. The first important clue for the basis of this tropism was the unexpected finding that a group of chemokines (RANTES, MIP-1α, and MIP-1β), small extracellular proteins naturally produced by CD8+ T cells, inhibited macrophage-tropic but not T-cell line–adapted strains.35 This discovery at once explained the nature of a long-sought CD8 viral suppressor factor and suggested a previously unexpected role for chemokine receptors. The independent identification of an orphan chemokine receptor, CXCR4, as the second receptor for T-cell line–tropic strains36 was followed by a rapid series of reports demonstrating CCR5 to be the principal second receptor for macrophage-tropic strains and CXCR4 for T-cell line–adapted strains.37–40 Crystallographic evidence indicates that CD4 binds in a recessed pocket on gp120, which includes a deep cavity that binds to phenylalanine-43 of CD4. Previous mutagenesis studies had shown this phenylalanine to be a crucial residue for binding.41 Other studies have also indicated a role for sugar molecules, glycosaminoglycans, in the binding of gp120, which influences interactions with the chemokine receptor.42 Binding to CD4 triggers a conformational change in gp120 that allows it to then bind to CCR5 or CXCR4. This second binding event exposes the fusion domain of the gp41 transmembrane protein, allowing fusion of the viral and cellular membranes, followed by viral entry into the target cell.
Events that occur immediately after viral entry—collectively, the disassembly process—are not simply the reverse of viral assembly. For example, studies have shown that HIV-1 must incorporate a cellular protein, cyclophilin A, which binds to the viral capsid protein p24. Failure to incorporate this cellular protein results in a profound postentry block during the next viral entry. Coincidentally, cyclophilin (peptidyl-prolyl isomerase) is the binding protein for cyclosporine, an inhibitor of T-cell activation, suggesting that activation-related cellular processes are involved in viral disassembly.43,44 In addition, virion infectivity factor (Vif) and “negative” regulatory factor (Nef; a misnomer), accessory viral proteins, may also be required.45
Reverse transcription begins in the cytoplasm, as DNA synthesis is initiated from the transfer RNA primer bound to the viral genomic RNA just downstream of the 5′ long terminal repeat (LTR). Reverse transcription proceeds in an orderly fashion in a similar manner in all retroviruses. Briefly, the transcription complex begins at the 5′ end, copies the U5 and R regions of the 5′ LTR, and then jumps to the 3′ end of the RNA, where the newly synthesized R region DNA binds to the R region of the 3′ LTR. Reverse transcription continues through the U3 region of the 3′ LTR and then through the remainder of the viral RNA, which gives a complete minus strand of DNA. The RNA is degraded by the viral ribonuclease H, except for two resistant purine-rich tracts in the middle and toward the 3′ end of the viral RNA. These then serve as the primers for formation of the DNA plus strand.46 Because reverse transcription takes place in the cytoplasm, local concentrations of nucleotides may be a limiting factor, particularly in nondividing cells. This is the rationale for using the ribonucleoside reductase inhibitor hydroxyurea to limit viral replication.47,48
During the formation of double-stranded DNA, the uncoated nucleoprotein complex, termed the preintegration complex, is imported into the nucleus.47,48 This is an energy-requiring process that uses nuclear localization signals present on viral Gag, viral protein R (Vpr), and integrase (IN) proteins. Unlike most retroviruses, which integrate into the host cellular DNA as the nuclear membrane is disrupted during cell division, HIV-1 can be imported into the nucleus and integrate into nondividing cells. This may be especially important in the infection of monocytes and macrophages, which are essentially nondividing cells.
IN-negative mutants of HIV do not integrate and do not produce infectious virus.49,50 Integration does not appear to be site directed. However, HIV preferentially integrates into or near active genes, particularly those that are activated after infection by HIV-1.51 Integration of the provirus appears to be an essential step in every replication cycle. Unintegrated viral DNA may survive, however, particularly in quiescent cells. This may provide a stable intermediate form in cells that are temporarily not permissive for infection; if cell activation occurs when these forms are present, viral infection may then proceed to completion. Integration of viral DNA establishes a linear copy of the viral genome within the genome of the cell, and replication of the virus then occurs along with cell replication. Integration is generally for the life of the cell and, with the cell and its progeny, for the life of the organism.
Synthesis of new viral RNA genomes and proteins is accomplished in a highly regulated manner by using host cell proteins. A high level of viral production from several different cellular compartments can be maintained throughout the course of infection. The high number of replication cycles allow the generation of variants and selection by drugs or the immune system. The half-life of virus-producing CD4+ T cells is approximately 0.7 day, and the generation time of HIV-1 in vivo is approximately 2 days.52 HIV-1 pathogenesis is the result of a complex interplay between the virus and the immune system, particularly the mechanisms responsible for T-cell homeostasis and regeneration. Protracted loss of CD4+ T cells results from early viral destruction of selected memory T-cell populations, followed by a combination of profound increases in overall memory T-cell turnover, damage to the thymus and other lymphoid tissues, and physiologic limitations in peripheral CD4+ T-cell renewal.53 Equally important are indirect hematopoietic and immunoregulatory effects of viral components (see later).
Once integration has occurred, virus production depends on the presence of cellular and viral factors required for activation of viral promoters. External factors, including coinfection with other agents, production of inflammatory cytokines, and cellular activation, may enhance viral replication.54 The molecular mechanisms regulating virus production include cellular pathways involving factors, such as the nuclear factor kappa B (NF-κB) family of inducible transcription factors that result in a cascade of events leading to viral genome expression.55
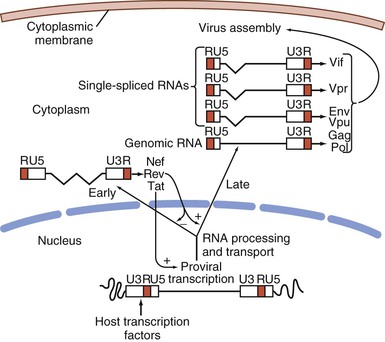
A unique feature of HIV-1 is that expression of different viral RNA species is temporally regulated. Using cellular enzymes, such as RNA polymerase II, transcription of the provirus is initiated at the viral promoter, at the junction of the U3-R regions in the LTR, as a single complete message. The viral messenger RNA (mRNA) and genomic RNA transcripts, processed by cellular machinery, are spliced, capped, polyadenylated, and transported to the cytoplasm for translation into viral proteins (see Fig. 171-2). Differential splicing of this complete RNA, controlled in part by the viral protein Rev, determines the type of message and protein that is produced. Early after infection, activated cells produce 2-kb mRNAs for viral regulatory proteins that can be detected by Northern blot analysis56; using even more sensitive reverse-transcription polymerase chain reaction (PCR) techniques, expression can be detected within 6 hours.57 These messages include the unique double-spliced RNA for trans-activator (Tat), Rev, and Nef proteins.58 Tat protein induces a markedly enhanced activity of the viral promoter, chiefly by facilitating the elongation of nascent short viral transcripts, resulting in greatly increased RNA and protein production. The Rev protein serves to decrease the production of double-spliced messages. With the accumulation of Rev protein, there is a switch to enhanced expression of unspliced and single-spliced mRNAs that encode the late viral proteins, including the virion proteins Gag, Pol, and Env and viral protein U (Vpu), Vpr, and Vif, as well as genomic RNA. The delayed transit from early to late viral genes probably reflects a requirement for threshold levels of Rev needed to bind and form multimers of the protein complexed with the Rev regulatory element (RRE), located in incompletely spliced mRNAs. Packaging of the genomic RNA within a virus particle requires the presence of a specific packaging signal located between the major splice donor at the 5′ end of the genome and the initiation codon for the Gag precursor polyprotein. In the absence of this signal, mature particles are formed that are devoid of RNA. The incorporation of genomic RNA requires two zinc-finger domains found in the p7 Gag nucleocapsid protein. Assembly of mature viral particles occurs at the cell membrane with the association of the Gag matrix protein p17 with the cytoplasmic domain of the envelope transmembrane protein gp41, which in turn binds to the viral gp120 on the outer surface. Assembled virions include the viral envelope proteins, cell membrane and associated cellular proteins, a matrix composed mainly of Gag p17, and a capsid composed of the p24 capsid protein and RNA, RT, IN, Gag proteins p6 and p7, and Vpr. The mature viral particle characteristically buds from the cell surface into the surrounding media, completing the life cycle of the virus. Budding of HIV-1 virions occurs at highly specialized membrane microdomains known as lipid rafts.59 These domains are characterized by a distinct lipid composition that includes high concentrations of cholesterol, sphingolipids, and glycolipids.
The Pathogen
The mature infectious virus particle buds from a cell membrane, forming a sphere with an outer lipid bilayer and a nucleocapsid with a dense, cone-shaped core (Fig. 171-3). The core appears to be attached to the viral outer envelope at its narrow end.60 The outer membrane contains up to 72 spiked knobs, which are assembled as trimers of the outer envelope protein gp120 bound to the transmembrane portion gp41. The viral membrane is cholesterol rich and includes cellular proteins.61

Each mature virion is composed of two molecules of single-stranded RNA surrounded by three gag gene cleavage products: the p17 matrix protein; the p24 major capsid protein, which forms the capsid shell; and the p7 nucleoprotein, which binds tightly to the viral RNA. The matrix contains the myristoylated matrix protein p17, which is critical for virion formation and is localized between the capsid protein p24 of the viral core and envelope. The p7 protein binds the two positive-strand copies of complete viral RNA attached at the packaging site, and it also binds to p24. A number of other viral proteins required for the early phases of infection are incorporated with the virion (Fig. 171-4): protease, which is essential for viral assembly; RT and IN, which are needed after entry for viral DNA synthesis and integration; transfer RNAlys (tRNAlys) at the 5′ end of the RNA, which serves as the primer for initiation of negative-strand DNA synthesis; and Vpr. The latter is a small protein that contains a nuclear localization signal and is associated with the nucleocapsid in large quantities. The virus encodes at least five other regulatory and/or accessory genes of diverse function. Some have been discussed already, and most are present in the infected cell but not in the mature virion. A list of HIV genes and associated proteins is presented in Table 171-1.

TABLE 171-1
Genes and Gene Products of HIV-1 and HIV-2
| GENE | PROTEINS | SIZE (kDa) | FUNCTION/PROPERTIES |
| gag | p17 | Matrix protein; interacts with gp41 | |
| p24 | Core protein | ||
| p6 | Core protein; binds to Vpr | ||
| p7 | Nucleocapsid; binds to RNA | ||
| P1 | |||
| p2 | |||
| pol | Protease | 10 | Proteolytic cleavage of Gag and Pol |
| Reverse transcriptase | 66, 51 | Polymerase and RNase H activity (p66 only) | |
| Integrase | 32 | Integration into chromosome | |
| env | gp120 | Envelope; viral entry into cell | |
| gp41 | Transmembrane protein; cell fusion | ||
| vif | Virion infectivity protein | 23 | Efficient cell-free transmission |
| vpr | Viral protein R | 18 | Enhances viral replication in primary cells, virion-associated protein; G2/M phase arrest; nuclear localization |
| tat | Trans-activator of transcription | 14 | Major viral trans-activator, immune suppression |
| rev | Regulator of expression of virion protein | 19 | Enhances expression of unspliced and single-spliced RNAs |
| vpu* | Viral protein U | 15-16 | Enhances virion release from cells; downregulates CD4 and MHC class I surface expression |
| nef | Negative regulatory factor | 27 | Inhibits or enhances viral replication depending on strain and cell type |
| Downregulates CD4; MHC class I | |||
| Antiapoptosis | |||
| vpx† | Virion protein X | 25 | Packaged into the virion |
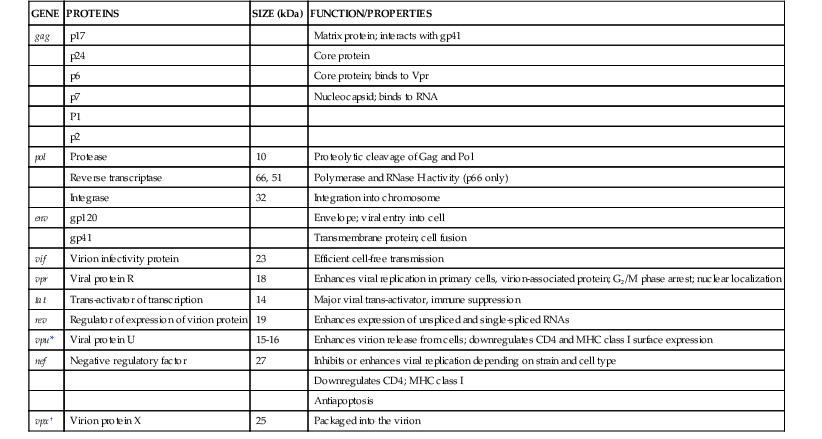
* HIV-1 only.
† HIV-2 only.
HIV, human immunodeficiency virus; MHC, major histocompatibility complex; RNA, ribonucleic acid; RNase H, ribonuclease H.
Genetic Organization
The HIV-1 proviral DNA integrated into the host cell is 9.7 kb in length and follows the basic genomic structure common to most retroviruses: gag-pol-env genes flanked by two complete viral LTRs (see Fig. 171-2). The provirus is symmetrically flanked at either end by the viral LTR and by cellular sequences representing the site of integration. These LTRs contain transcriptional regulatory sequences, RNA processing signals, packaging sites, and the integration sites. The 5′ end begins with the gag gene, which encodes core and matrix proteins; the pol gene, which begins in an overlapping frame encoding viral protease, RT, and IN; and then the env gene, which encodes the outer and transmembrane envelope proteins. A complex series of open reading frames encode the accessory proteins. vif is partly contained within the pol coding region and partly downstream, and vpr is located downstream from vif. The first coding exons for tat and rev are co-linear and located between vpr and env, and their second exons, located in the env gene, are joined to the first exons by RNA splicing. The vpu gene is co-linear with the 5′ region of the env gene; nef is located downstream from the env gene and extends into the downstream LTR sequence.
Transcription of a single unspliced RNA is initiated at the 5′ end by the cellular RNA polymerase II. The unspliced mRNA serves as a template for translation of the Gag and Gag-Pol precursor polypeptides. This message is also spliced to produce single-spliced transcripts for Vif, Vpr, and Vpu proteins and the Env precursor polypeptide, and double-spliced transcripts for Tat, Rev, and Nef. The precursor polyproteins are then cleaved by cellular or viral enzymes; the Gag and Gag-Pol precursors are cleaved by the viral protease, which is itself transcribed from the unspliced viral message. The Env precursor polypeptide is cleaved by cellular proteases (Fig. 171-5).
Virion Structural Proteins
Gag Proteins
The cleavage of the Gag precursor protein by viral protease produces the structural components of the viral core and matrix. To form the virus capsid structure, a large polyprotein is made from the viral mRNA. This 55-kDa Gag precursor protein (sometimes called p55) is cleaved into at least five structural proteins by the viral 34-kb protease encoded at the 5′ end of the pol gene. Lack of protease function, either through inhibition by drugs or after transfection of the p55 gene into a cell that lacks protease, results in the formation of noninfectious viral particles.62
The p55 protein is seen on Western blot preparations made from whole-cell lysates but not on those made from mature virions. During or shortly after self-assembly, the viral protease is activated, and the precursor is cleaved into three principal proteins and two smaller peptides. These proteins undergo extensive post-translational modification by cellular enzymes. After translation, the initiating methionine residue is removed, p17 is myristoylated, p17 and p24 are phosphorylated by cellular kinases, and p7 (the nucleocapsid protein, or NC) binds to two zinc ions to form the zinc fingers that bind to RNA. Although the virus depends on cellular systems for many functions, it requires the virally encoded protease for the cleavage of the Gag proteins. Gag proteins are sufficient to form particles when expressed from transfected cells. These self-assembled particles are noninfectious. The p17 matrix protein, MA (molecular weight, 17,000), contains about 130 amino acids and is myristoylated on a glycine at its amino terminus by the host cell enzyme N-myristoyl transferase.63,64 The first 31 amino acids target the myristoylated protein to the cell membrane, and nonmyristoylated proteins cannot form infectious virions. In addition to viral assembly at the cell surface, MA functions as part of the preintegration complex targeting viral DNA to the cell nucleus and enabling HIV-1 to infect and integrate into nondividing cells such as macrophages.65 p17 has extracellular functions via binding to the IL-8 receptors CXCR-1 and CXCR-2 expressed on peripheral blood mononuclear cells (PBMCs)66 and increases their production of proinflammatory cytokines and counteracts the inhibitory activity of IL-4 on these cells.67
The p24 capsid protein, CA, is produced by two cleavages to form a 240–amino-acid hydrophobic protein that forms the major subunits of the viral capsid and self-associates to form dimers and higher-order structures. This protein binds the cellular cyclophilins, a process that may be important for viral replication.68 A major homology region of 29 amino acids is shared with many retroviruses.69,70 p24 Gag is typically the easiest protein to detect using sera from infected patients, and serologic detection in retrovirus-infected animals gave the name group antigen (Gag) to these proteins. The carboxyl-terminal sequences encode a 70–amino-acid hydrophilic protein, NC, that binds both viral RNA and the capsid p24 protein, intertwining approximately one molecule with four to six nucleotides of RNA,71 and another small protein, p6. The p7 zinc-finger domain binds RNA and recognizes the packaging site on Gag.72,73 The p6 protein appears to mediate the incorporation of Vpr into the virion and facilitates release of the virion from the cell surface, perhaps because of its binding to tumor susceptibility gene 101 (Tsg101) and ALIX (ALG2-interacting protein X), two cellular budding factors.74 Two small peptides of unknown function, p2 and p1, are also found in the viral core.
An intracellular antiviral mechanism targets the capsid protein and is mediated by the tripartite motif (TRIM) family.75 The TRIM motif consists of a RING (really interesting new gene) domain at the N-terminus, a B box-2 domain, and a “coiled coil” domain,76 and it apparently represents a widespread and ancient innate host defense mechanism. TRIM5α appears to be functionally similar to the Fv1 restriction element, which regulates permissivity of murine cells of different genetic backgrounds for infection with different strains of tropic murine leukemia viruses (MuLVs), and was identified 40 years ago.77 The restriction by both human TRIM5α and murine Fv1-B of N-tropic MuLV infection depends on residue 110 of the MuLV capsid protein.78 HIV-1, however, is only partially sensitive to human TRIM5α (depending on allelic variants),79 perhaps helping to account for its successful cross-species transmission from chimpanzees, although macaque TRIM5α strongly restricts HIV-1.75,78 TRIM5α binds to viral capsid hexamers80 and inhibits replication at a postentry step. The precise mechanism is not clear and may be mediated at more than one level, but may dysregulate viral capsid uncoating.80,81
Viral Enzymes
pol Gene Products
The pol gene encodes three enzymes, protease, RT, and IN. These proteins are synthesized from the same mRNA as the Gag proteins through a ribosomal translational frameshift. The cleavage of the 160-kDa precursor polyprotein is essential for viability. Because of the inefficiency of the frameshift, which is important in regulating the relative levels of viral proteins, there are about 2000 copies of each Gag protein and only 100 copies of each Pol protein per virion.
Protease
Protease is a 10-kDa 99–amino-acid protein that is fully active as a dimer. It is autocatalytically cleaved from the precursor protein during the viral assembly process. Site-specific mutagenesis has demonstrated that noninfectious particles containing uncleaved Gag and Gag-Pol proteins are produced if this enzyme is inactivated. The similarity of viral protease to other aspartyl proteases, such as angiotensin-converting enzyme, has greatly facilitated the design of potent antiviral drugs, including inhibitors of dimerization and molecules that bind to the active catalytic site.62,82
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


