INTRODUCTION
The human immunodeficiency virus (HIV) epidemic is as modern an epidemic as it is large. This epidemic has advanced with the cultural and technological transformations of the 20th century. However, research, treatment, and (one hopes) a cure have also proceeded incredibly rapidly, as the full force of modern science was turned on the pandemic.
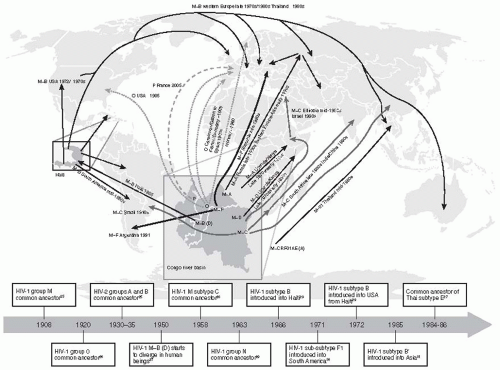
A zoonotic infection of humans occurring in Congo in the 1890s, HIV infection simmered slowly in Africa, manifested by increases in cryptococcal meningitis and disseminated tuberculosis. However, these were always common infections in Africa. Later, modern transportation allowed the infection to spread across the continent. Serological evidence of HIV infection was found in stored serum samples from subjects from Zaire that had been collected in 1959.
1 The probable origin of HIV-1 was from
Pan troglodytes (chimpanzees) in West and Central Africa, with the infection then being transmitted to humans during hunting for “bush meat.”
2 Another human immunodeficiency virus, HIV-2, was discovered subsequently in African green monkeys in the wild in West Africa.
3 Humans can be infected either by HIV-1 or HIV-2, and both viruses can cause immunodeficiency and acquired immunodeficiency syndrome (AIDS), although the prevalence and virulence of HIV-2 are considerably less than those associated with HIV-1.
4 and
5 From there, airline travel took the virus to Haiti, as this island nation has close cultural ties to West Africa. From Haiti, it spread to popular holiday destinations in the Dominican Republic. In particular, the Dominican Republic was a preferred holiday destination for the increasingly visible and sexually active community of men who have sex with men (MSM). The sexual revolution of the late 1960s and 1970s created an ideal environment for the virus to spread.
AIDS, as it became known, is the severe state of immune depression caused by years of HIV infection. AIDS was first identified in the United States in 1980-1981 among MSM. Pentamadine availability was managed by the Centers for Disease Control and Prevention (CDC), as it was a rarely used drug for
Pneumocystis pneumonia treatment in children whose immune systems were suppressed by cancer chemotherapy. When CDC received multiple requests for pentamadine to treat adult men, the cluster of illness was noted. Early epidemiology studies identified clusters of
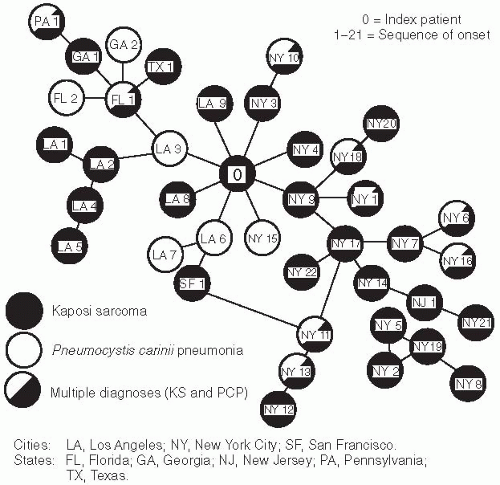
Pneumocystis carinii pneumonia (PCP) and Kaposi’s sarcoma (KS) (or both) occurring in MSM in Los Angeles, New York, and a few other cities, many of whom had reported sexual contact with another case (
Figure 22-1).
6,
7,
8 and
9 Subsequently, similar patterns of disease were noted among injection drug users, persons with hemophilia, and some transfusion recipients.
10,
11Several research groups investigated the causes of the immunosuppression underlying the occurrence of these opportunistic infections. In 1984, Robert Gallo and colleagues at the National Cancer Institute of the National Institutes of Health (NIH) and Luc Montagnier and coworkers at the Pasteur Institute independently reported the discovery of the cause of AIDS, a novel human retrovirus, the human immunodeficiency virus.
12,
13
THE HIV VIRUS
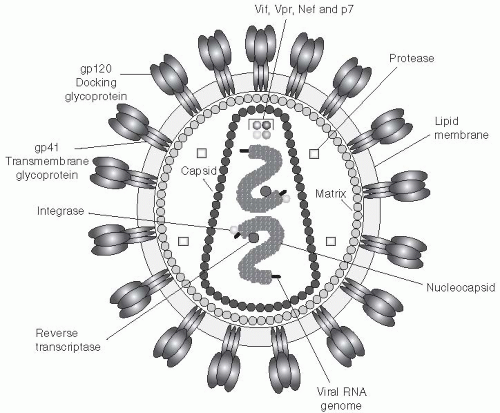
HIV is a retrovirus, a double-stranded RNA virus that undergoes reverse transcription to form doublestranded DNA in the cytoplasm of an infected cell (
Figure 22-2). The viral genome consists of three structural genes, termed
env, pol, and
gag. The genome codes for several regulatory proteins, including Tat, Rev, Vif, Vpu, Vpr, and Nef. The p16/p14 Tat proteins activate viral transcription. The p14 Rev protein is responsible for the transport and stability of viral RNA. The p27/p25 Nef proteins are active in the downregulation of CD4 cells.
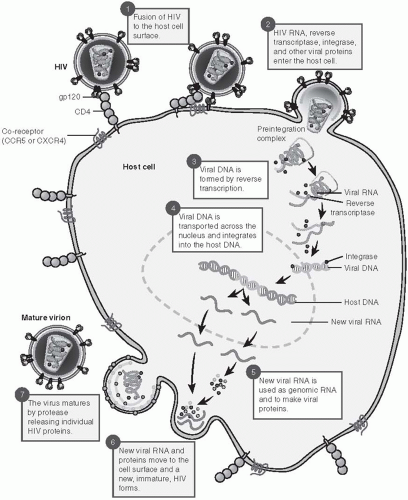
15The HIV virus attaches to the CD4 cell surface molecule through high-affinity interactions between the viral envelope protein, Gp120, and a specific region of the CD4 molecule (
Figure 22-3). The CD4 molecule is present in abundance on both immature T lymphocytes and mature CD4+ helper T lymphocytes. It is also present in lower concentrations on monocytes, macrophages, and antigen-presenting dendritic cells. HIV also binds to one of two co-receptors, CCR5 or CXCR4. Once the CD4 molecule and the co-receptor are engaged, the viral attachment complex shifts conformation, thereby allowing the membrane fusion complex to fuse with the cell wall and the viral capsid to enter the cell. Cells of monocyte/macrophage origin generally express only CCR5; however, many lymphocyte populations express both receptors.
After the virus enters the cell, the viral RNA is converted to DNA using the viral enzyme reverse transcriptase. The double-stranded DNA molecules then enter the nucleus and are integrated into the host cell chromosomal DNA. In resting cells, the transcription and integration may be incomplete and viral DNA may remain in the cytoplasm. The unintegrated DNA is short-lived; in contrast, integrated DNA can persist for long periods in resting T lymphocytes.
16Subsequently, transcription of the HIV genome occurs to form RNA transcripts, which leave the nucleus. Modification of the viral polyproteins by cellular and viral enzymes (proteases) is essential for viral assembly. The virus is then spliced and repackaged near the cell surface and released from the host cell.
HIV has a high replication rate, with approximately 10 billion viral particles produced each day.
16,
17 As an RNA virus, it does not undergo RNA polymerase editing, and the reverse transcriptase results in a huge error rate in viral replication. It has been estimated that every day every possible amino acid is mutated in the production of so many viral particles. The result is a swarm of viable and non-viable viruses causing high levels of immune system activation, but with only a fraction of the viruses actually being able to productively infect other cells. Although HIV itself is not long-lived (its half-life is estimated at 6 hours), the genome is integrated into both activated immune cells and incredibly long-lived central memory T cells. These long-lived cells serve as the viral reservoir and enable the virus to become reactivated even after decades of effective antiretroviral therapy.
18
HIV NATURAL HISTORY
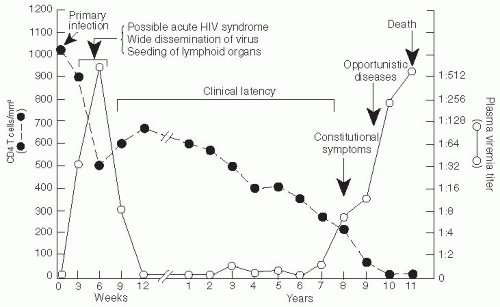
After HIV infection either by sexual contact or parenterally, the virus is present in the blood and the viral RNA can be detected within 7-10 days. Approximately 7-21 days later, HIV antibodies appear in the blood. Early in the infection, the viral load is usually very high.
19 Infection of gut-associated lymphoid tissue (GALT) occurs rapidly, and the destruction of GALT is complete within the first few weeks of infection. Subsequently an immune response occurs, which includes the appearance of antibodies and a cellular response consisting of CD8+ cytotoxic T lymphocytes directed against the virus. At this point, the level of viremia declines and a viral “set point” is established in 3-4 months (
Figure 22-4). The level of the viral set point can vary considerably and is predictive of the rate at which the disease will progress. The AIDS mortality among untreated persons with very high viral load at the set point—that is, more than 100,000 copies/mL—is quite rapid and averages about 4.5 years, whereas persons with lower viral loads at the set point survive more than 10 years after their infection.
20After the set point is reached, a gradual decline in the level of CD4+ T lymphocytes occurs along with an increase in the level of CD8+ T lymphocytes, so that the total number of CD3+ T cells is relatively stable for several years. Nevertheless, the CD4/CD8 lymphocyte ratio is significantly decreased (i.e., less than 1.0) compared to the uninfected state. During this time, the body experiences an ongoing, destructive war between the immune system and the virus. Massive numbers of viral particles are produced, and the immune system responds to their presence. Activated immune cells are susceptible to infection, so the very response to the virus accelerates infection of additional cells. Cells traffic to the lymph nodes to perform immune surveillance, but the lymph architecture is increasingly scarred by the severity of the immune response: as fibrosis increases, the lymph nodes become less and less able to perform their vital functions.
21Approximately 18-24 months prior to the development of clinical AIDS, the level of total CD3+ cells decreases more rapidly, meaning there is loss of T-cell homeostasis.
22 The loss of T-cell homeostasis heralds the patient’s inability to keep up with the virus-induced destruction of the immune system. This phenomenon is often accompanied by a “switch” in the co-receptor utilization of the dominant HIV, from a CCR5, or nonsyncitium-inducing cell type, to a CXCR-4 (syncitiuminducing or duotropic) type. Rarely, CXCR-4 viruses are transmitted initially and predominate even early in the infection; in such cases, the progression to clinical AIDS occurs more rapidly (
Figure 22-5).
23During the acute stage, which occurs 2-4 weeks after the initial infection when HIV levels in the blood are maximal, an “acute HIV infection syndrome” commonly occurs; this phenomenon has been reported in approximately 50% or more of patients in some closely observed cohorts in developed countries.
24,
25 and
26 The symptoms noted during this time resemble infectious mononucleosis and include fever (95%), adenopathy (75%), pharyngitis (70%), rash (70%), and other systemic symptoms, including meningitis, Guillain-Barré syndrome, peripheral neuropathy, and Bell’s palsy.
24,
25 and
26 Generally, HIV
serology is negative during this acute seroconversion syndrome, but patients have demonstrable viremia. Seroconversion occurs within a few weeks after the prodromal phase resolves.
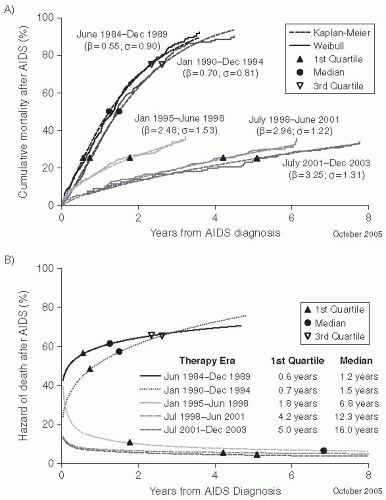
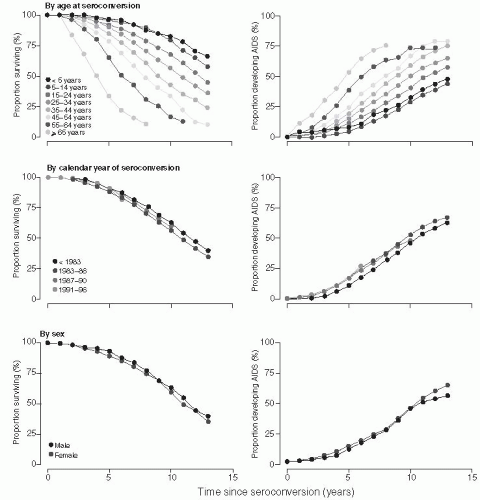
A meta-analysis of the time from HIV-1 seroconversion to AIDS and death (before the widespread use of highly active antiretroviral therapy [HAART]) among 13,030 HIV-1 seroconverters enrolled in 38 studies in Europe, North America, and Australia— the CASCADE study
27—has provided the most comprehensive evaluation of the time to AIDS and death among persons in developed countries in the pre-HAART era. The CASCADE investigators found that the median time to AIDS was 9.5-11.0 years, and the median survival was 10.5-11.8 years after infection in untreated patients (
Figure 22-6). There were no significant differences in progression by exposure category, gender, or year of infection between 1983 and 1996. However, the age of the person at the time of infection substantially affected the progression rate. Persons who were older when they were infected progressed more rapidly. Among persons aged 15-24 years at the time of infection, the median survival was 12.5 years (95% confidence interval [CI]: 12.0-12.9 years), and the median time to AIDS was 12.0 years (95% CI: 10.7-11.7 years). In contrast, among persons 45-54 years old at seroconversion, the median AIDSfree interval was 7.7 years (95% CI: 7.1-8.6 years) and the median survival was 11.0 years (95% CI: 10.7-11.7 years). After it became possible to quantitate HIV viral RNA in the plasma, it was shown that the levels of virus in the plasma at the set point were predictive of the subsequent rate of HIV progression and could be combined with the CD4+ count to more accurately predict the time of onset of AIDS and death in HIV-infected patients.
19,
20 In fact, the combination of the viral RNA (i.e., viral load) with the CD4+ count more accurately predicts the time to AIDS than the CD4+ count alone.
20 This relationship reflects how the CD4 count indicates the degree of immunosuppression, and the viral load indicates the level of immune control versus viral replication and pathogenesis.
Fewer data are available on the rate of progression among persons in developing countries. Because death is the result of uncontrolled infections, the exposure of patients to pathogens is a key element of survival. In areas of the world characterized by a higher prevalence of aggressive infectious diseases, such as tuberculosis or untreated cancer, life expectancy is much shorter. A study in Thailand found that progression was more rapid among young adults compared to HIV-infected persons of comparable age in developed industrialized countries, with median survival of 7.4-8.4 years after seroconversion among infected Thais.
28
AIDS-RELATED OPPORTUNISTIC INFECTIONS
The diagnostic hallmark of untreated HIV is the development of AIDS and its accompanying opportunistic infections (OIs). A large number of OIs have been identified in immunosuppressed HIVinfected persons and have been classified as “AIDSdefining illnesses.” The CDC lists 28 conditions as “AIDS-defining illnesses.” In the United States,
Pneumocystis carinii pneumonia, the HIV wasting syndrome, Kaposi’s sarcoma, oropharyngeal and esophageal candidiasis, extrapulmonary cryptococcus, and tuberculosis account for most initial AIDSdefining OIs in patients (
Table 22-1).
The frequency of the initial AIDS-defining diagnosis among cases reported to the CDC in 1990, 1995, and 1997, using the pre-1993 clinical definition, is shown in
Table 22-1. AIDS-related OIs in developing countries often have a different distribution at the onset of AIDS than has been reported among patients in the United States and other industrialized countries. In Thailand, for example, the distribution of the top 10 AIDS-defining illnesses among 101,945 cases of symptomatic AIDS that were reported between 1994 and 1998 is shown in
Table 22-2.
29Thailand is similar to many other developing countries in Asia and Africa in that tuberculosis is the most frequent AIDS-defining illness, followed closely by the wasting syndrome and several systemic fungal infections—namely, candidiasis, cryptococcosis, and penicilliosis. Also, PCP, which has been classified as a fungal infection caused by
Pneumocystis jiroveci (formerly known as
P. carinii), is common in Thailand. The frequent occurrence of PCP differs from the clinical reports of AIDS in Africa, where PCP among adults has been reported to be rare, although it appears to occur commonly in pediatric AIDS cases.
30 Infection with
Penicillium marneffei is limited to AIDS patients in Southeast Asia, where the organism is endemic and is geographically localized to this area.
31 Kaposi’s sarcoma is relatively rare among AIDS patients in Asia but is quite common among such patients in Africa and the Caribbean.
29,
32 Disseminated histoplasmosis is a common AIDS-defining illness among patients in the histoplasmosis belt in the U.S. Midwest.
33 Leishmaniasis is a common AIDS-related OI in Spain and other areas where this organism is endemic.
34
Timing of AIDS-Related Opportunistic Infections
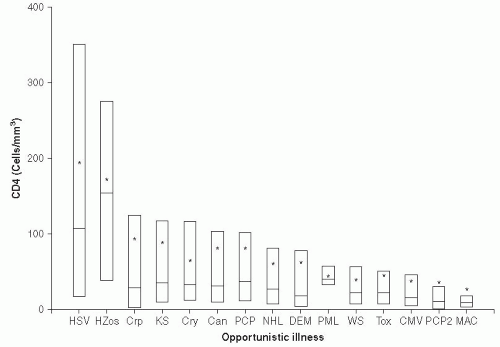
The time of occurrence of AIDS-related opportunistic infections during the natural history of HIV infection varies with each OI (
Figure 22-7). Typically, oropharyngeal and esophageal candidiasis, oral hairy-cell leukoplakia, and tuberculosis occur relatively early in an HIV infection, when the CD4+ count may be 200-300 cells/µL or higher. Disseminated cytomegalovirus infections (CMV), toxoplasmosis, and
Mycobacterium avium infections usually do not occur until the CD4+ cell count falls below 50-100 cell/µL. This knowledge of the natural history of HIV has been used to develop antibiotic protocols to prevent frequently occurring OIs in patients with HIV. In some cases, antibiotic prophylaxis of an opportunistic infection has been shown to delay the progression of HIV/AIDS and to prolong survival of HIV-positive patients. Prolonged survival has been shown for prophylaxis of PCP,
tuberculosis, and
M. avium infections.
35 It is equally likely (although no clear data have been reported) that prevention of fungal infections may prolong survival of antiretroviral-naive patients (
Table 22-3).
Because of increasing availability of antiretroviral therapy (ART), emphasis has shifted to treatment of the underlying immune suppression with effective ART rather than prophylaxis in immunesuppressed patients. Unfortunately, not all patients will effectively respond to ART, and many will still need prophylactic regimens to prevent illness. A CDC task force has reviewed the published literature and the available scientific evidence and published recommendations for the use of prophylactic regimens for the prevention of specific AIDS-related OIs.
35 Immune reconstitution following ART is also not complete; while CD4 cell counts may rise to levels that would seem sufficient, some patients have immune deficiency previously associated with lower absolute T-cell counts. Thus these patients require closer clinical follow-up.
HOST FACTORS IN SUSCEPTIBILITY OR RESISTANCE TO HIV INFECTION AND DISEASE PROGRESSION
After infection with HIV, the host mounts an immune response consisting of both cellular and humoral immunity. This response usually results in some control of viral replication with a substantial decrease in the viral load.
Because HIV transmission rates are low, an effective immune response—either innate or adaptive—must be mounted against the virus in many exposed people. Innate antiviral factors and physical characteristics of the body surface repel the virus in most cases. Mucosal IgA and IgG antibodies and CD8+ T cells may inhibit HIV at the mucosal surface and prevent or clear HIV infection after exposure. Also, CD8+ and CD4+ T cells may clear infection after
the virus has penetrated the mucosal barrier early in infection. Clearance of virus may occur prior to the appearance of neutralizing antibodies; however, exposed persons may harbor T lymphocytes that respond specifically to HIV peptides by proliferation and cytokine secretion.
36,
37 Documentation of these events is difficult and highly disputed in HIV transmission research circles. HIV dogma states that infection is irreversible and that there are no cases of viral clearance after infection. However, acquired immunity after HIV exposure has been demonstrated among frequently exposed persons— for example, in a cohort of Kenyan commercial sex workers—and nonprogression of the infection for prolonged periods has been observed in other infected persons.These so-called “elite controllers” of their HIV infection may have normal levels of CD4+ T lymphocytes and undetectable HIV RNA while not receiving specific antiretroviral therapy for many years. These findings give rise to optimism that these protective mechanisms could be enhanced to produce a sterilizing or pathogenesis-controlling vaccine.
In addition to the acquired immune response, several genetic host factors have been discovered that influence resistance or susceptibility to infection. Among these host factors is a common genetic mutation involving the deletion of 32 base pairs in the chemokine receptor CCR5. Persons who are homozygous for this mutation are resistant to infection with CCR5-utilizing viruses.
38 Persons who are heterozygous for the deletion have lower viral loads and slower progression to AIDS.
39 In 2007, an HIV-positive U.S. citizen living in Germany, Timothy Brown, developed leukemia and required a bone marrow transplant. Because the German healthcare system has a more generous system for picking an ideal match, his physician, G. Hütter, was able to search for a donor candidate who was both an HLA match and had the CCR5 deletion (donor 61). Brown stopped taking HIV medication before his bone marrow transplant and has not had detectable virus since the transplant. He did suffer a relapse of his leukemia, but received a second transplant from the same donor and has remained in remission. He is apparently cured of his HIV infection. Numerous questions remain in this case. In particular, why the virus completely disappeared is unclear. Despite aggressive ablative therapy prior to the transplant, this patient still has CCR5-carrying cells in his body, and no amount of radiation would eliminate all the immune cells. Furthermore, the patient had CXCR4 viruses detected prior to the transplant, and these viruses have not emerged. This result has energized the search for a cure to HIV infection.
40Additional host genetic factors have been identified that affect the resistance or susceptibility to HIV-1 infection or the rate of progression to AIDS after infection. These include a mutation in the CCR5 promoter, a CCR2-641 mutation, and a mutation in the stromal-derived factor (SDF13’A/3’A).
41,
42 The number of segmental duplications of two CC chemokine genes, CCL3L1 and CCL4L1, on chromosome 17q were recently shown to be associated with the susceptibility to HIV infection and the rate of progression after infection in several human populations.
43Persons who are homozygous at one, two, or three HLA class 1 loci or have a polymorphism for interleukin 10 (IL-10+/5’A or 5’A/5’A) have more rapid progression after they are infected with HIV.
44 However, homozygosity of class 1 HLA alleles has not been shown to increase the risk of HIV infection. In contrast, HLA class 1 allele sharing between an infected person and an exposed seronegative recipient (through either a sexual contact or a mother-infant pair) has been shown to increase the risk of transmission.
45,
46 Also, one study has found discordance of an HLA class II allele, HLA-DRB3, to be protective for the sexual transmission of HIV-1 among a sample of couples.
47Specific HLA alleles have also been shown to influence the genetic susceptibility to HIV-1 infection. The alleles HLA-B*27 and B*57 have been consistently associated with a favorable prognosis after infection, mostly by influencing early viral equilibration,
44 although they do not protect against HIV infection. In contrast, HLA-B*35 and HLA-B*53 have been associated with an unfavorable prognosis and higher viral load in infected individuals.
44 To date, no other single HLA-B allele has been shown to have a significant influence on the disease progression, nor have any associations between HLA-A or HLA-C alleles and natural history been demonstrated convincingly. However, research in the genetic influences on HIV susceptibility and natural history is being pursued by several research groups at present.
The combined effect of several of these genetic host factors on the natural history of HIV infection has been studied among 525 homosexual men in the Multicenter AIDS Cohort Study cohort who seroconverted between 1984 and 1996, prior to the advent of HAART.
48 On the basis of a regression tree analysis using a Cox proportional hazard model for times to AIDS, it was estimated that 30% of the men enrolled in MACS had one or more genetic resistance factors. The participants with the genetic resistance determinants experienced slower
progression early after their infection, although the effect was limited to the first four years after infection. Clearly, additional evaluation of host resistance factors influencing HIV infection and progression will be important. Such studies require large cohorts of carefully followed subjects in various populations in whom genetic diversity can be evaluated. The Center for HIV AIDS Vaccine Immunology (CHAVI) is an example of one of a large collaboration of geneticists and other basic scientists working to bring together the wealth of genetic samples to answer these key questions in HIV vaccinology and cure. Some of the host factors currently believed to be related to susceptibility or resistance to HIV-1 infection and progression are listed in
Table 22-4.
Research has discovered several cellular factors that suppress HIV-1 replication. It has been known for some time that several human and primate cell lines were naturally resistant to infection with HIV-1. One mechanism of this natural resistance has been identified as being due to a protein called tripartite motif (TRIM) 5-alpha, which has ubiquiton ligase activity. When the TRIM 5-alpha gene from monkey cells that were resistant to HIV-1 infection was transferred into and expressed by normally susceptible human cells, resistance to HIV-1 infection was induced.
49 This finding could eventually prove important for the development of new therapeutic approaches to control HIV-1 infection.
Another cellular mechanism to defend against infection with HIV-1 and other retroviruses has been described. This resistance is mediated by a cytidine deaminase termed APOBEC3G.
50 APOBEC3G mutates deoxycytidine to deoxyuridine in the negative strand of viral DNA; these multiple mutations render the virus inactive. However, many retroviruses, including HIV, have evolved the
vif (viral infectivity factor) gene to inactivate APOBEC3G. The successful replication of HIV-1 in cells is determined by a balance between cellular APOBEC3G and
vif. Recently, important mutations in APOBEC3G have been discovered.
51 One such mutation, H186 RR, has been associated with a more rapid decline in T lymphocytes and progression to AIDS. This codon-changing variant gene was found to be common among African Americans (
f = 37%) but is rare in European Americans (
f = 5%) with HIV-1 infection.
52Researchers continue to study genetic polymorphisms that influence HIV infection, transmission, and risk of progression. Despite the information gained through extensive genetic exploration, however, it is likely that HIV is affected by a broad constellation of host genes interacting with viral genes. This complex interaction between multiple genes complicates efforts to understand all the relevant interactions.
INFLAMMATION AND MICROBIAL TRANSLOCATION
As HIV-infected patients are now living longer thanks to effective therapy to suppress their HIV, their risk for chronic diseases is of increasing concern. Diabetes, cardiovascular disease, liver and kidney failure, and cancer are the leading causes of morbidity and mortality among this population. Not only are the rates of these diseases elevated compared to HIVnegative persons, but there is also an indication they may occur at younger ages. Given this pattern, HIV has been postulated to be a model of accelerated aging. It is not yet clear if this increase in morbidity and mortality is a direct result of the viral infection and the immune response or to drug toxicity.
Research is ongoing to investigate immune activation or inflammation as the underlying cause of these pathologies. For instance, cardiovascular disease is now recognized to be the result of inflammation that causes macrophages to traffic into arterial walls, form foamy cells, and eventually create plaques, which can subsequently rupture. Evaluation of inflammation markers among HIV-positive patients has found increased levels of chronic inflammation markers, including C-reactive Protein (hsCRP), D-dimer, interleukins 6 and 10 (IL-6 and IL-10), and many other biological markers.
The underlying cause of this inflammation remains under study as well. The infection with loss of gut-associated T lymphocytes (GALT) in early infection disrupts gut mucosal integrity and results in elevated plasma levels of bioactive bacterial lipopolysaccharide (LPS) and bacterial DNA—specifically, conserved sequences of DNA coding the 16s rRNA subunit (16s DNA). This increase leads to chronic inflammation and depletion of CD4+ T memory cells. This pathogenetic sequence of events, which is termed microbial translocation, is only partially reversed by effective antiviral therapy. Even in patients with well-controlled viremia, some replication is ongoing, which may also contribute to inflammation. Attempts to eliminate the last reservoirs of virus with intensification regimens and the addition of anti-inflammatory drugs to the antiviral regimen are under study.
53
HIV GENOTYPES
HIV-1 has been classified into three groups, based on genetic relatedness—namely, groups M, N, and O. The latter two groups are geographically limited to countries in West Africa. Group M strains of HIV-1 have been divided into 11 genetic subtypes, designated A-K. Particular subtypes are found more commonly in certain areas of the world. In the Americas and Europe, subtype B strains have predominated, whereas in Africa various subtypes have been more common in different countries. In India, subtype C has predominated, and in Thailand and other countries in Southeast Asia, subtype E (now called CRF01_AE) has predominated.
Recombinants are becoming more common over time, and genetic subtypes are showing significant dynamism between and across countries, risk groups, and regions. Data from Africa, Asia, and South America have shown that a significant percentage of circulating strains represent mosaics of two or more subtypes. Coinfection by different subtypes is apparently not uncommon, leading to the conclusion that infection with HIV does not protect individuals from reinfection. Packaging of RNA from different subtypes into the same viral particle, coupled with the strand-switching activity of reverse transcriptase, generates recombinant HIV in coinfected individuals. Since full-length viral sequencing became more efficient in the past 5 years, it has become clear that subtypes and recombinants are equally important in the pandemic. Recombinant viruses are now identified as circulating recombinant forms (CRFs), if the same recombinants are commonly found in different geographic regions, or unique recombinant forms (URFs), if they are found in only a few individuals. The distribution of HIV-1 genotypes in three East African countries (Uganda, Tanzania, and Kenya) has been found to include 35-45% URFs, emphasizing the frequency of coinfections or super-infections with different HIV-1 genotypes in these populations.
54 Subtype E has been classified as a recombinant subtype since the first full-genome sequences were done; several other strains have been recognized as recombinants based on full-length or partial sequences.
54 HIV is a rapidly changing virus and the original subtypes can be imagined to be snapshots of the virus at the time they were defined and not actual specific types that are maintained due to some unknown selective advantage.
The relationship between HIV subtypes and clinical progression has been investigated in several populations with diverse strains. For example, in more than 2000 hospitalized patients in Thailand, the level of immune suppression, spectrum of opportunistic infections, and mortality were similar with subtype B-infected and CRF01_AE-infected patients.
55 However, a study of survival after infection with subtype CRF01_AE strains among Thai military conscripts found significantly shorter median
survival (7.8-8.4 years) than has been reported among cohorts of persons from Western countries infected with subtype B strains (9.5-11 years).
56 Whether the decreased survival was due to viral, host, or environmental factors is not clear. In Kenya, where subtypes A, C, and D cocirculate, the plasma RNA levels were highest and the CD4 counts were lowest in persons infected with subtype C viruses.
57 In Uganda, subtype D infection has been reported to be associated with more rapid progression than subtype A.
58 In contrast, a study in Sweden showed no differences in the rate of CD4 cell decline or clinical progression among persons infected with subtypes A, B, C, or D.
59 In a prospective study in Senegal, persons infected with subtypes C, D, or G were eight times more likely to develop AIDS during follow-up than those infected with subtype A.
60 It is difficult to determine whether the apparent variation in progression among persons infected with different subtypes is due to genetic differences in the viruses, host characteristics, or other factors.
Studies of the genetic diversity of HIV strains have proved very useful in tracing epidemiologic patterns of HIV-1 transmission in populations. Among injection drug users (IDUs) in Russia, several subtypes of HIV-1—namely, subtype A, subtype B, and a recombinant A/B strain—have demonstrated drug trafficking links between Kaliningrad, a Russian enclave on the Baltic Sea
61 and more distant areas of Russia. Similarly, four overland heroin-trafficking routes in southeast Asia have been defined in this manner (
Figure 22-7).
62
IMPACT OF COINFECTIONS ON HIV
It is now widely accepted that the progression to AIDS among HIV-1-infected individuals is highly correlated with HIV-1 viral load and the CD4 cell count.
63,
64 It is also likely that differential immune activation is the primary driver of the variability in progression to AIDS in persons infected with HIV-1.
65,
66 Over the last several years, evidence has accumulated in support of the theory that coinfections, through chronic immune activation and its consequent effect on host immunity and HIV progression, may account for the enhanced HIV infection rate, accelerated progression of disease, and reduced survival seen in sub-Saharan Africa.
67
Tuberculosis
Worldwide, tuberculosis (TB) is the most serious opportunistic infection among HIV-infected individuals. Previous advances to control the disease were eliminated by the HIV epidemic, allowing for a resurgence of tuberculosis.
68 HIV has also fueled the current epidemics of multidrug- and extremely drug-resistant TB.
Tuberculosis most often comes under immunologic control in an HIV-positive patient and remains latent. There is an elevated risk of disease in the first year after infection, which then drops substantially to yield a 10% lifetime risk of active disease. The dominant cytokine response in
M. tuberculosis infection is Th1, and 90% of individuals newly infected with
M. tuberculosis do not develop active TB, in part, due to the Th1 immune response. However, when there is a Th1/Th2 shift, the predominant Th2 response may predispose such individuals to an increased risk of both HIV and active
M. tuberculosis when they are exposed.
69 HIV-infected patients are then at high risk of active disease (10%) in the first year, and face an additional 2% risk each year that they will progress to active disease. Patients are also at higher risk of acquiring TB again. HIV-positive persons living in high TB incidence areas are also at an extremely high risk of TB, TB activation, and TB reinfection. Complicating this picture is the fact that TB diagnosis in HIV-positive patients is more difficult, as these individuals are often smear negative and, therefore, may be falsely screened as negative.
Because the TB and HIV epidemics have substantial geographic overlap, control of each disease is dependent on control of the other. Treatment of HIV before advanced immune suppression, treatment of TB for a year, and prophylaxis of patients with isoniazid (INH) are necessary to control the disease. Development of novel TB drugs that will shorten treatment times in HIV-positive patients and point-of-care diagnostic tools are active areas of research.
Treatment of Patients Who Are Coinfected with Tuberculosis and HIV
Patients with AIDS frequently are diagnosed when they have active, often disseminated, tuberculosis. Despite the availability of effective therapy for tuberculosis, it is the most common cause of death in AIDS patients in South Africa. The optimal timing of antituberculous chemotherapy and highly active antiretroviral therapy (HAART) was unclear until recently because of the common appearance of immune reconstitution syndrome (IRIS) in such patients during HAART treatment and drug interactions between rifampin and some classes of antiretroviral drugs. Also, the high pill burden, overlapping side effects, and programmatic difficulties
in monitoring and managing therapy were common challenges.
To define the optimal course of therapy for patients coinfected with HIV and tuberculosis, a randomized controlled trial was done in South Africa, known as the Starting Antiretroviral Therapy at Three Points in Tuberculosis (SAPIT) trial. In this study, 642 patients were enrolled to evaluate the outcome among those who were treated with integrated TB and anti-HIV therapy compared to those in whom therapy was sequential, with TB therapy given first, followed by anti-HIV treatment. This trial found that mortality was 56% lower (5.4/100 person-years) when antiretroviral therapy began soon after TB therapy than when antiretroviral therapy was delayed until the completion of TB therapy (12.1/100 person-years). It is now recommended that HAART therapy be initiated soon after initiating TB therapy—for example, 4-6 weeks, after beginning anti-TB therapy in coinfected patients.
70
Helminths
It has been hypothesized that parasitic infections— specifically, infections with helminths (schistosomiasis, ascaris, bookworm, and taenia)—may be responsible, in part, for the increased susceptibility and spread of HIV in the developing world.
71 Specifically, the immune activation caused by helminthic infections is thought to increase the susceptibility to and progression of HIV in coinfected persons. The evidence supporting this hypothesis comes in part from studies of Ethiopian Jews who have emigrated to Israel, as well as residents of the Western Cape region of South Africa. The majority of Ethiopian immigrants were infected with helminths, and their immunological profiles were dominated by activation of a Th2 response.
71,
72,
73 and
74 Some evidence indicates that Th2 clones are more permissive for HIV-1 infection, and a Th2 predominance or a Th2 immune activation may impede the development of antiviral immunity.
73 This immune activation is also marked by increased expression of HIV co-receptors and decreased secretion of β-chemokines in vitro.
73 Further, studies in some populations have found that anti-helminthic treatment has been associated with decreased HIV plasma viral load. However, the reduction in HIV viral load after anti-helminthic therapy has not been observed in all African populations.
75
Malaria
Together, malaria and HIV cause more than 4 million deaths per year, with more than 90% of these deaths occurring in sub-Saharan Africa. These highly disease-burdened regions overlap geographically, prompting further speculation about the possibility of a direct interaction between HIV and malaria.
Early studies failed to find a significant direct impact of malaria on the prevalence or progression of HIV.
76 However, children with severe anemia due to malaria are at risk of transfusion-transmitted HIV. With the creation of the World Health Organization’s (WHO) Roll Back Malaria partnership in 1998, as well as increased funding for both of these diseases from the Global Fund for HIV, Tuberculosis, and Malaria, increased attention has been paid to these three significant diseases. The possibility of interactions between HIV and malaria has major public health implications, and the growing body of evidence lending support to this possibility justifies the need to better understand these interactions (
Table 22-5). Recent studies have demonstrated an increased HIV-1 viral load in persons with malaria, which decreased with successful treatment of malaria.
77 Studies of pregnant women have found an increased rate of mother-to-child transmission (MTCT) of HIV-1 associated with placental malaria.
78 A study in Uganda of patients with partial immunity to malaria found that HIV-1-infected persons more frequently developed clinical malaria and had higher parasite levels than those who were HIV negative.
79
Hepatitis C Virus
Because of shared routes of transmission, hepatitis C virus (HCV) infection is common in HIV-positive individuals and is considered an opportunistic infection of HIV.
80,
81 and
82 In the United States, 15-30% of HIVinfected persons are also infected with HCV; however, the prevalence of HIV-HCV coinfection varies markedly by the route of acquisition of HIV infection (risk category). For example, among HIV-positive patients seen at the Johns Hopkins HIV Clinic (
n = 1955), HCV coinfection prevalence rates were as follows: 85.1% of those reporting injection drug use, 14.3% of those reporting heterosexual contact, and 9.8% of those reporting male homosexual contact.
78HCV infection in patients with HIV infection has been associated with higher HCV RNA viral load and accelerated progression of HCV-related liver disease.
81,
82 and
83 One study reported that HCV RNA levels were higher in persons with hemophilia who became infected with HIV than in those who did not, and that liver failure occurred exclusively in those coinfected with HIV and HCV.
83 In a study of liver disease and hepatocellular carcinoma among 4865 men exposed to HCV-contaminated blood products, at all ages the risk for liver-related death after HCV
exposure was 1.4% and 6.5% for HIV-negative and HIV-positive individuals, respectively.
84 Data from most studies confirm the detrimental impact of HIV infection on hepatitis C infection. As survival of HIVinfected persons has been extended due to HAART and the prophylaxis of traditional OIs, it is likely that hepatitis C morbidity and mortality will increase.
85A significant challenge in HCV research has been the difficulty encountered when trying to measure the state of liver pathology. Although serum markers of liver function have been developed to evaluate the functional capacity of the liver, they have considerable variability and their predictive value is more complex in dual infection. Liver biopsy remains the gold standard, but it is an invasive procedure that measures the status of the liver only in the location of the biopsy. One new technique that shows promise in evaluating the entire liver is an ultrasound scan of the liver, known as fiberscan. This device is licensed in Europe, but as of 2012 had not yet secured U.S. approval. If approved by the U.S. Food and Drug Administration (FDA), it provide give a less invasive measure of liver function across the entire organ.
The issue of whether HCV infection also adversely affects progression of HIV disease remains controversial.
82 In a prospective study of 416 HIV seroconverters in Italy, those with and without HCV infection progressed to AIDS at similar rates.
86 Among the 1955 men seen at the Johns Hopkins HIV Clinic, there was no difference in progression to AIDS or death associated with HCV infection after adjusting for HAART and HIV suppression.
79 Conversely, one study of 3111 persons receiving HAART reported that HCV-infected persons had a modestly increased risk for progression to a new AIDS-defining event or death, even among the subgroup with continuous suppression of HIV replication.
87 This same study found that CD4 increases were smaller after effective anti-HIV therapy in persons with HCV infection than in those without such coinfection. However, a review of the literature—including two subsequent studies evaluating the immunologic response to potent antiretroviral therapy—failed to confirm these observations.
88,
89 and
90Recent advances in HCV therapy have resulted in significantly higher cure rates for all HCV-infected patients. These treatments raise the possibility that many coinfected patients can clear their HCV infection. To date, uptake of HCV therapy has been slow, but is gaining momentum. Advances in the next few years may change the HCV/HIV epidemic considerably.
GB Virus C
GB virus C (GBV-C) is a flavivirus that is closely related to hepatitis C virus. Although GBV-C was first detected by nucleic acid amplification in patients with non-A, non-B hepatitis, the virus does not replicate in hepatocytes and is not a hepatitis virus. In fact, GBV-C has not been associated consistently with any human disease (it is not even screened from the U.S. blood supply), although infections are quite common among injection drug users and homosexual men.
GBV-C replicates in CD4+ T cells.
91 Numerous studies have shown no effect or a beneficial effect for patients coinfected with GBV-C and HIV.
92,
93,
94,
95,
96,
97,
98,
99,
100 and
101 Most of these studies were done among cohorts of prevalent HIV-1-infected persons, in whom the duration of HIV-1 infection was not precisely known but was estimated by modeling.
87,
88,
89,
90,
91,
92,
93,
94,
95,
96,
97 and
98 The impact of GBV-C on untreated HIV required a study with known date of HIV acquisition.
The longstanding Multicenter AIDS Cohort Study (MACS) has known dates of seroconversion. In this cohort, it was found that there was no effect of GBV-C on survival when it was measured shortly after seroconversion; when measured 5 to 6 years after HIV seroconversion, however, men without GBV-C viremia were 2.78 times more likely to die than men with persistent GBV-C infection. Men in the MACS cohort who cleared their GBV-C viremia had more rapid progression and poorer survival than either those with persistent infection or HIV-positive men who never experienced a GBV-C coinfection.
102At the same time, a report from the Amsterdam cohort of homosexual men found no association between GBV-C persistent viremia and survival or progression to AIDS; instead, men who lost their GBV-C viremia were three times more likely to progress than men who never had GBV-C virus infection in their cohort.
101 These authors postulated that loss of GBV-C viremia was a marker for loss of sufficient CD4+ T cells to support continued GBV-C replication. Data were reported recently from the Viral Activation Transfusion Study in which patients with advanced HIV infection who needed transfusions were randomized to receive either leukoreduced or nonleukoreduced red cell transfusions. The mortality was 78% lower (HR = 0.22) among patients who acquired incident GBV-C infections in this study despite similar CD-4+ cell counts and HIV viral load at baseline prior to their GBV-C incident infection.
374 These prospective data were free of the bias associated with higher GBV-C infection prevalence in subjects with higher CD-4+ cells in several other observational studies.
These studies highlight the importance of understanding the full longitudinal history of a disease. Without multiple samples to evaluate changes in GBV-C status over time, the apparent inconsistencies in the relationship could not have been elucidated. Interest in GBV-C has waned considerably as the protective effect of the coinfection has been demonstrated to be substantially weaker than the effect of ART. Previous protocols to intentionally infect participants with GBV-C who have limited drug options due to resistance have not been pursued as new drug classes have been licensed.
Laboratory research into mechanisms of viral interference of GBV-C has continued. In vitro experiments have found that GBV-C-infected CD4+ T cells are more resistant to infection with HIV-1.
103 Also, the expression of a gene for the chemokines RANTES, MIP 1-α, MIP-1-β, and SDF-1 and secretion of the chemokines into the culture supernates were higher in GBV-C-infected cells. Surface expression of CCR5 was also significantly lower in GBV-C-infected cells.
103
Other Agents
Several other coinfections have been reported to be associated with transient decreases in HIV-1 viral load, including measles,
104,
105 dengue fever,
106 and scrub typhus.
107 The mechanism of the suppression of HIV replication during these acute infections is believed to be associated with immune activation and elevated cytokine and/or chemokine levels from the coinfection. However, these acute infections are associated with only transient reductions in the viral load. In addition some investigators have reported HIV-2 co-infections to be associated with slower progression and lower viral load among patients with HIV-1 infections.
375
ANTIRETROVIRAL THERAPY
No Therapy to Monotherapy/Dual Therapy
HIV therapy can be loosely divided into several time periods. The pre-therapy period was from zoonosis until 1983, when the disease was recognized. Treatment of opportunistic infections and prophylaxis for OIs was the only option until AZT’s licensure. From 1987 until 1996, a few more nucleoside reverse transcriptase inhibitors (NRTIs) came to the market; thus this era is called the “monotherapy” or “dual-therapy” era.
There has been a marked improvement in the natural history of HIV infections when responses are measured at the population level. An evaluation of the proportion of men in the Multicenter AIDS Cohort Study who were receiving various forms of therapy between 1986 and 1999 and their progression to AIDS documented the benefits of various regimens in a cohort of HIV-infected men. A comparison of the progression of infection among HIV-positive MACS participants, after adjusting for the duration of infection at baseline, found the relative hazard of progression to AIDS during the era of no treatment to be 1.52 (95% CI: 0.93-2.49) compared with men in the monotherapy era.
108 The hazard of progression in the era of combination therapy was 1.03 (95% CI: 0.77-1.38), and during the era of HAART it has been 0.31 (95% CI: 0.21-0.45).
108Mortality from AIDS in the general population in the United States declined dramatically with the availability of HAART after 1996. A CDC-sponsored HIV outpatient study (HOPS) reported the use of HAART therapy and mortality among 1000 patients with AIDS
and CD4+ cell counts less than 100 cells/mm
3. In these patients, approximately 80% were using protease inhibitors (PIs) by 1997, and the mortality declined from 29.4 deaths per 100 person-years in 1995 to 8.8 deaths per 100 person-years in 1997 (
Figure 22-8). In addition to decreased mortality, the incidence of AIDSdefining opportunistic infections have been reduced dramatically in the HAART era.
109HIV therapy is in constant evolution and the reader interested in the latest recommendation should go to Internet sources to find the most up-to-date recommendations. There are also several excellent references that are frequently updated, including
The Johns Hopkins Hospital 2012 Edition, Medical Management of HIV infection, by John G. Bartlett and Joel E. Gallant (hopkins-aids.edu/mmhiv/order.html.) In addition, the International Antiviral Society-USA panel makes updated recommendations for therapy on a regular basis. Their recommendations are published in the Journal of the American Medical Association.
378As a retrovirus, HIV has numerous pathways that are not used by the human host and, therefore, are good drug targets. The first antiretroviral to show any success in clinical trials was zidovudine (AZT). AZT is an NRTI that was developed in cooperation between Burroughs Wellcome and the National Cancer Institute out of cancer therapy programs that had been interested in retroviruses as causes of cancer. AIDS activism and the Ronald Reagan administration’s drive to reduce federal regulation synergistically pushed for an accelerated approval process so that AZT moved from the laboratory to approval in a little more than two years. AZT was approved for use in HIV patients on March 20, 1987, and as a postexposure prophylaxis in the 1990s. Early optimism, which emerged as patients were rescued literally from death’s door, was
crushed by HIV’s ability to rapidly develop resistance to this therapy. To effectively treat this rapidly mutating virus, it became clear, a multipronged treatment approach would be necessary. In the early 1990s the protease inhibitor class began clinical trials, and on December 6, 1995, saquinavir was approved.
Hit Early, Hit Hard
While many patients received HIV medications prior to licensure because they were enrolled in clinical trials and received drugs under “compassionate use” clauses, the highly active antiretroviral therapy (HAART)—in Europe, called combination antiretroviral therapy (CART)—era began in earnest in 1996. Most patients were able to suppress the virus to levels below the threshold of detectability with the licensed viral load assays (i.e., fewer than 50 copies/µL). This led to the hypothesis that with aggressive therapy for several years, the infection might be “cured” through the combined effects of the drugs and the host’s immune system. The catchphrase for HAART during this period became “Hit hard, hit HIV early.”
110Subsequently, the latent viral reservoir in long-lived resting lymphocytes and macrophages was described, leading to a change in thinking about appropriate antiretroviral therapy. Modeling the time to the natural elimination of this reservoir of latent virus suggested that the reservoir of these cells or their daughter cells would persist for 60 years or more.
111 Adding to this discovery of the latent reservoir was the recognition that patients who were treated for several years with repeatedly undetectable viral loads commonly returned to their pretreatment viral loads within a few weeks after withdrawal of treatment. This observation led to the rejection of the hypothesis that HIV infection could be cured with currently available drugs.
Unfortunately, the new drug regimens had severe toxicities. Complications of such therapy include a variety of conditions, especially lactic acidosis, lipodystrophy, visceral fat accumulation, insulin resistance, and diabetes, especially with thymidine analogs, such as d4T (stavudine), AZT (zidovudine), and protease inhibitors. Also, peripheral neuropathy, hepatitis, rash (including Stevens-Johnson syndrome), and renal toxicity have been reported as complications of therapy with antiretroviral drugs. The reader should consult references describing HIV/AIDS therapy for more details.
Treatment-Sparing Regimens
Because of the cost of antiretroviral therapy, the pill burden, and the side effects, it was considered possible to give intermittent treatment relief to patients whose virus was suppressed and in whom the CD4+ cell count had recovered. In addition, prospective studies determined that many patients taking antiretroviral drugs often interrupted their therapy. Therefore, it was proposed to purposefully interrupt therapy in selected patients who had controlled their HIV infection, in a practice called “structured treatment interruption.”
To determine whether structured treatment interruptions were associated with adverse outcomes, two large clinical trials were conducted. The first trial, called Strategies for Management of Antiretroviral Therapy (SMART), enrolled 5472 patients. In 2720 of these individuals, therapy was interrupted when the CD4+ cell count exceeded 350 cells/mm
3 and restarted after it fell below 250 cells/mm
3 (the treatmentsparing arm of the trial). In the control group of 2752 patients, therapy was continued uninterrupted (the viral suppression arm). The study ended early in January 2006 when it was found that opportunistic disease or death occurred in 120 patients in the treatment-sparing group (3.3 events per 100 personyears) and in 47 patients in the viral suppression group (1.3 events per 100 person-years).
112In the other trial, known as the Staccato trial, 450 subjects with CD4+ cell counts greater than 350 cells/mm
3 and HIV viral load less than 50 copies/mL were randomized to receive either continuous treatment or interrupted therapy. This smaller study also found that adverse events were more common in those patients with interrupted therapy, although they experienced cost savings and their HIV viruses did not become resistant to drugs.
113After the results of the SMART trial were reported, an expert committee of the International AIDS Society recommended against treatment interruptions.
114
Viral Suppression and Cure
Today, supported by studies that demonstrate the lasting damage the HIV virus has on the immune system, and facilitated by new, more convenient regimens with better toxicity profiles, the pendulum has swung back to earlier treatment. Some sources even advocate treatment at the time of infection to preserve as much immune function as possible. In fact, the most recent recommendations of the Expert Committee on Antiretroviral Therapy of the International AIDS Society are to consider offering treatment to any patient with HIV infection despite the CD-4+ cell count .
The North American AIDS Cohort Collaboration Observational Research Database (NA-ACCORD) study reported on the risk of AIDS and death among patients starting ART in Canada and the United States. Using a marginal structural method (MSM) analysis, these researchers evaluated survival according to the CD4 cell count at which 17,571 participants
initiated ART. MSM analysis allows the study team to structure the data as though patients were entering a hypothetical clinical trial in which patients who started with between 500 and 351 CD4 cells/mm
3 were compared to those who started fewer than 350 cells/mm
3. Those who started at the lower CD4 cell count were found to have a 69% increased risk of death. In a second analysis, those who initiated therapy with fewer than 500 CD4 cells/mm
3compared to those who started therapy with higher CD4+ cell counts had a 94% higher risk of death.
114The HIV Causal Collaboration gathered data on more than 20,000 patients in the United States and Europe. In a more robust analysis using a dynamic marginal structural model pioneered by Hernan and Robins, this team modeled a hypothetical clinical trial in which multiple CD4 strata were defined and patients were assigned a status of “initiated” or “deferred” within each strata. The team found that the risk of AIDS or death was increased by 38% for those who delayed treatment until after their CD4 cell counts fell below 350 cells/mm
3 and by 90% for those who delayed treatment until their CD4 cell counts fell below 200 cells/mm
3.
115These novel statistical methods have been difficult to explain to the broader medical community, but the consistency of the finding that AIDS and death are reduced when ART is initiated earlier is reassuring. As these methods are further refined and strengthened, it is likely that observational data will increasingly be used to inform clinical effectiveness.
Now that the virus can be well controlled with antiviral medications, emphasis has shifted to finding “the cure.” A cure for HIV could be achieved either by completing eliminating the virus or by developing a robust immune response that could control the virus in a latent state indefinitely.
Immune Reconstitution Disease
A particularly devastating side effect of HIV/AIDS treatment is immune reconstitution inflammatory syndrome (IRIS). Patients in whom effective antiretroviral therapy is begun, and particularly those who were severely immunosuppressed with CD4+ T-cell counts below 100 cells/mm
3, can develop disseminated immune reactions. As the functioning immune system is restored and the CD4+ T-cell count increases, symptoms often
appear from an inflammatory response to an infection that was previously tolerated during the time of severe immunosuppression.
117 Signs and symptoms of IRIS include fever, pleural effusions, ascites, pulmonary infiltrates, osteomyelitis, meningitis, renal failure, and occasionally mortality. IRIS most commonly occurs in patients who are coinfected with HIV and TB.
The first reports of
M. tuberculosis-associated IRIS were published in 1998 after the advent of HAART.
118 Since them, IRIS has been reported in
M. avium-infected patients,
119 patients infected with
M. leprae or
M. kansasii,120 and patients who received bacillus Calmette-Guerin (BCG) vaccine when they were immunosuppressed.
121 This syndrome has also been seen with a range of other opportunistic infections, including cytomegalovirus, hepatitis B and C viruses,
Cryptococcus neoformans, Pneumocystis jarovecki/carinii progressive multifocal leukoencephalopathy (caused by JC virus), leishmaniasis, and cerebral toxoplasmosis. Control of severe IRIS reactions can usually be managed with the use of corticosteroids, in
addition to specific anti-infection therapy directed at the opportunistic pathogen.
122
 cells/mm3
cells/mm3