Chapter 52 Human Herpesvirus-6 and Herpesvirus-7 Infections
In 1986 a novel virus was isolated from six patients with lymphoproliferative syndromes, two of whom were also infected with the human immunodeficiency virus (HIV).1 Molecular and structural characterization of the new virus revealed an icosahedral core structure of 162 capsomeres, indicating that it was a herpesvirus. Because of the initial belief that the new virus selectively infected freshly isolated human B cells, the virus was given the name human B-lymphotropic virus.1 Subsequent investigation revealed a broader cell tropism, with notable T-cell lymphotropism.2 For this reason, current nomenclature refers to this virus as human herpesvirus-6 (HHV-6).
Four years after the discovery of HHV-6, Frenkel and colleagues isolated another novel virus from the CD4+ T lymphocytes of a healthy adult.3 Designated human herpesvirus-7 (HHV-7), this new virus is closely related to HHV-6,4 exhibiting partial antigenic cross-reactivity with HHV-6.5,6 In 1994 DNA from the eighth member of the human herpesvirus family was isolated from Kaposi sarcoma lesions of HIV-infected individuals. Human herpesvirus-8, or Kaposi sarcoma-associated herpesvirus, is discussed in Chapter 51.
HUMAN HERPESVIRUS-6
Since its discovery in AIDS patients, HHV-6 has been implicated as a possible pathogen in HIV-infected persons or a cofactor in HIV disease progression. As researchers investigated and characterized HHV-6 in HIV-infected adults, Yamanishi and colleagues pursued its association with disease in immunocompetent children, discovering in 1988 that HHV-6 caused the common childhood disease exanthem subitum (roseola).7 As with all herpesviruses, HHV-6 establishes latency following primary infection during childhood, and reactivation from latency can result in active viral replication in both immunocompromised and immunocompetent persons.
Virology
Classification
HHV-6 is a member of the Herpesviridae family. Genomic analysis places HHV-6 among the β-herpesviruses, along with human cytomegalovirus (CMV) and HHV-7. Similarities with respect to amino acid sequences, gene organization, and putative protein functions suggest that HHV-6is closely related to both CMV and HHV-7.3,8,9 On the basis of DNA restriction analysis, in vitro tropism studies, and antigenic relations defined by reactivities of monoclonal antibodies, HHV-6 can be separated into two variants: variant A (HHV-6A) and variant B (HHV-6B).10 Characteristic HHV-6A isolates include GS (the original isolate) and U 1102 (isolated from a Ugandan AIDS patient).1,11 The prototypic HHV-6B isolate is Z 29, isolated from a Zairian AIDS patient.12 The intravariant nucleotide sequence homology ranges from 97% to 100% and the intervariant homology from 94% to 96%.13,14
Viral Composition
Virion Structure
An enveloped virus, HHV-6 has an icosahedral nucleocapsid consisting of 162 capsomeres.15 The capsid diameter measures 95–105 nm, with the diameter of the nucleoid being 60–80 nm. The nucleocapsid is surrounded by a dense, prominent tegument 25–40 nm thick.15 Extracellular enveloped virions range from 160 to 200 nm, comparable to that of herpes simplex virus.
Genomic Structure and Genetic Content
Contained within the nucleocapsid of HHV-6 is a linear, double-stranded DNA molecule of 160–162 kb (Fig. 52-1). The genome consists of a 143–145 kb unique sequence (U) flanked by direct repeats, DRL (left) and DRR (right), of 8–9 kb and interrupted by three intermediate repeats (R1, R2, and R3) in the immediate-early A (IE-A) region.16,17 The variation in length is due to the heterogeneous (het) region at the left end of both DR elements.18,19 The genomic organization of the U region of HHV-6 is similar to that of the unique long (UL) region of CMV and is co-linear to that of HHV-7. The mean G + C content is 43%.8,16,18 The complete nucleotide sequences of HHV-6A (U 1102) and of HHV-6B (Z29 and HST) have been reported.16,17,20 The genomes contain 119 distinct open reading frames likely to encode proteins.20 Nine open reading frames of HHV-6A do not have counterparts in HHV-6B.16 The genes in the U region are termed U1 to U100 and open reading frames within the direct repeats are designated DR1 to DR7, whereas the nine open reading frames unique to HHV-6B are termed B1 to B9.21 The overall nucleotide sequence identity between HHV-6A and HHV-6B is 90%.20
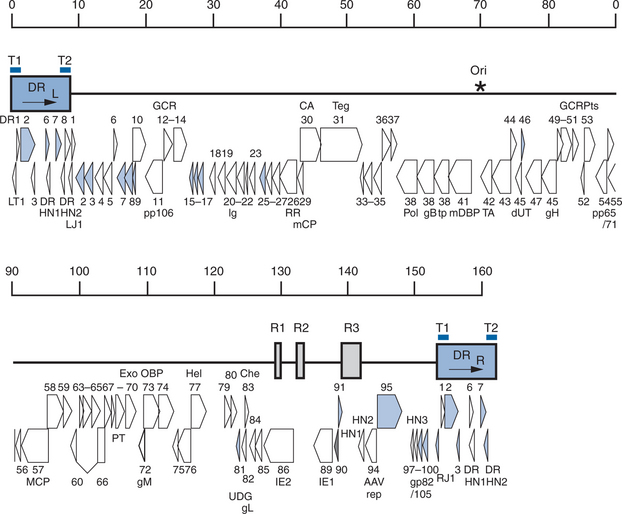
In Vitro Biologic Properties
HHV-6 exhibits predominantly CD4+ T-lymphocyte tropism.2,24 It can be isolated from CD4+ CD82 and CD31 CD4+ mature T lymphocytes but not from CD4+ CD8+, CD42 CD82, or CD32 T cells.24 Additionally, HHV-6 can be propagated in CD4+ CD8+, CD4+ CD82, and CD31 CD4+ cells with mature phenotypes and rarely in CD4+ CD8+ human cord blood mononuclear cells (CBMCs).24 CD46 serves as the receptor for HHV-6,25 and is present on the membrane of all nucleated cells. For HHV-6A, the complex of glycoprotein H (encoded by the HHV-6 gene U48), glycoprotein L (encoded by the HHV-6 gene U82), and glycoprotein Q (encoded by the HHV-6 gene U100) is the viral ligand for human CD46.26 Glycoprotein H is the component responsible for binding at the short consensus repeat domains SCR2 and SCR3 of CD46.27 The HIV-1 receptors and co-receptors CD4, CXCR4, and CCR5 are not receptors for HHV-6.28,29
Consistent with the ubiquity of the CD46 receptor, HHV-6 infects a multitude of tissues in the body, including brain,30–32 liver,33–35 tonsils,36 salivary glands,37 and endothelium.38 At the organ level, co-infection with HHV-6 and HHV-7 results in increased quantities of HHV-6 viral genome.39 Likewise, reactivation of HHV-6 by infection with HHV-7 has been achieved in vitro.40
Efficient HHV-6 replication in primary cell culture requires both prior mitogen activation of primary T cells, as provided by phytohemagglutinin (PHA),1,12 and full progression of the cell cycle, as demonstrated by the requirement for interleukin-2 (IL-2).41 Mitogenic anti-CD3 monoclonal antibodies can also enhance HHV-6 replication.42
The HHV-6A and HHV-6B strains have both been adapted by serial passage to replicate in continuous cell lines. Established human cell lines that support HHV-6A replication include those of T cell (including the T-lymphoblastoid cell line HSB-2), B-cell, megakaryocyte, and glial cell lineages, as well as transformed cervical epithelial cells.19 HHV-6B strains replicate well in the Molt-3 T cell line and the T-cell lymphoma line MT-4.41,43
Epidemiology
Geographic Distribution
Seroprevalence studies of HHV-6 infection have demonstrated remarkable reproducibility from widely separated regions of the globe. With rare exceptions,44 the prevalence of antibodies to HHV-6 is high among populations throughout the world.
Incidence and Prevalence of Infection
Epidemiologic studies in normal children have shown that most primary HHV-6 infections occur within the first year of life.45,46 HHV-6 immunoglobulin G (IgG) can be detected in more than 90% of neonates,47,48 reflecting both the high seroprevalence of HHV-6 among adults48,49 and the active transport of HHV-6 IgG across the placenta.46 The prevalence of HHV-6 IgG drops significantly by 4–6 months of life as maternal antibodies decline, then increases through the third year of life and remains high into adulthood.45,46,48 The highest geometric mean titers of HHV-6 antibody occur during the first 3 years of life, indicating a predominant clustering of primary infections in infants and toddlers.46,49 More than 90% of normal children become infected with HHV-6 by 12 months of life,48 and virtually 100% acquire infection by 3 years of age.49
Transmission
Although the mode(s) of transmission of HHV-6 has yet to be proven definitively, most children probably acquire infection through contact with the secretions of adult caretakers shedding the virus in saliva.37,50–52 Reports of culture isolation of HHV-6 from the saliva of healthy adults50,52 and adult patients infected with HIV52 document salivary shedding in more than 85% of persons. Viral shedding in saliva is intermittent,53 and the serum antibody titer to HHV-6 does not correlate with the ability to isolate HHV-6 from saliva samples.52 HHV-6 DNA can be detected by polymerase chain reaction (PCR) in saliva or PBMCs of more than 90% of healthy individuals.54,55 Using in situ hybridization and immunohistochemical staining, HHV-6 DNA and HHV-6 protein expression have been demonstrated in tissue from submandibular glands,37 parotid glands,37 salivary glands,51 and bronchial glands.51 Although breast milk is unlikely to be an important source of early HHV-6 infection,56 several reports suggest that HHV-6 may infect an infant congenitally or perinatally.57–60 Definitive proof of such transmission, however, remains elusive.
Pathogenesis
Viral Replication and Latency
Analysis of pathologic specimens reveals that HHV-6 can infect a wide variety of cell and tissue types. During acute infection, infection of lymphocytes, macrophages, histiocytes, endothelial cells, and epithelial cells occurs, with CD4+ T lymphocytes being the predominant target cell type in the blood. The cell target of HHV-6 in the oropharynx during primary infection remains under investigation. Persistent HHV-6 infection can occur in salivary glands and brain tissue,30,31,37,61 and HHV-6 has been shown to establish latency in mononuclear cells62,63 and early bone marrow progenitor cells.64 Low concentrations of HHV-6 DNA can be found in PBMCs from healthy individuals.
Immune Responses to HHV-6 Infection
Humoral Immune Response
The protective effect of antibody is suggested by the epidemiology of HHV-6 infection. As noted above, passively acquired maternal antibody wanes over the first 6 months of infancy; and during the next 6 months of life the number of children acquiring HHV-6 for the first time approaches 90%.48 This finding suggests that maternal antibody provides protection against primary HHV-6 infection. Additionally, during primary HHV-6 infection the clearance of viremia coincides with the appearance of HHV-6B-specific neutralizing antibody,65 again suggesting the importance of the humoral arm of the immune response to HHV-6.
Following primary HHV-6 infection, IgM-neutralizing antibodies appear within 5–7 days. Maximum IgM titers appear at 2–3 weeks, and the titers then decline and reach undetectable levels after 2 months.66 Host IgG responses following primary HHV-6 infection develop 7–10 days following the febrile period.67 Serologic evidence of previous infection manifested by HHV-6 IgG is maintained indefinitely.
Cellular Immune Response
The T-cell immune responses against HHV-6 infection are readily identified in healthy adult populations, and HHV-6-specific T-cell clones have been isolated.68,69 Moreover, the importance of the cellular component of the host immune response in maintaining latency is suggested by the increased frequency of HHV-6 reactivation in solid organ transplant recipients.70 Complete understanding of the role of cellular immunity in primary HHV-6 infection and in viral reactivation from latency awaits further investigation.
Clinical Relevance of HHV-6
The only disease for which HHV-6B has been shown definitively to be the causative agent is exanthem subitum (roseola), a common disease of childhood.7,67 In addition, HHV-6B is a major cause of emergency room visits and hospitalizations for infants and young children.58 HHV-6 produces a spectrum of neurologic diseases as well, including encephalitis and febrile seizures.71–73
Clinical Relevance in Relation to HIV Infection
In Vitro Data
Because HHV-6 was initially isolated from two HIV-positive patients and exhibits CD4+ cell tropism, it was hypothesized that HHV-6 may act as a cofactor in the acceleration of HIV infection to AIDS in patients co-infected with the two viruses. Initial evaluation of possible interactions between HHV-6 and HIV led to the discovery that the viruses can simultaneously infect the same CD4+ T lymphocytes under experimental conditions.2,74 Studies evaluating the impact of HHV-6 co-infection on active HIV viral replication in vitro have yielded contradictory results, with some investigations documenting enhanced HIV replication,74–76 whereas others reported inhibition of HIV replication.12,77–80 HHV-6 infection in the presence of HIV tat results in significantly higher HIV long terminal repeat (LTR) activation than that observed by tat or HHV-6 alone, indicating that HHV-6 and tat interact synergistically.81,82 Effects of co-infection on HHV-6 replication are less impressive: HHV-6 replication is unaffected78,80 or slightly inhibited77 in cells co-infected with HIV. However, expression of HIV-1 tat inhibits HHV-6 replication more dramatically, as shown by a 3.6- to 15.4-fold reduction in yield of infectious virus.81
HHV-6 induces expression of CD4 receptor molecules on the cell membranes of infected lymphocytes, hematopoietic progenitor cell lines, natural killer cells, and lymphoid tissue ex vivo, rendering them susceptible to co-infection with HIV.83–89 In vitro studies demonstrate that such co-infection leads to accelerated cellular apoptosis.74,85 Transactivation of the CD4 promoter by the IE1 and IE2 proteins of HHV-6 is responsible for this enhanced CD4 receptor expression.90 HHV-6 has been shown to downregulate the HIV-1 co-receptor CXCR4 in vitro,29,91 although expression of both CXCR4 and CCR5 are not affected by HHV-6 in an ex vivo propagation system.92 HHV-6 replication enhances the production of the CCR5 ligand RANTES,89,92,93 thus providing a mechanism for the selective blockade of CCR5-tropic but not CXCR4-tropic HIV-1 replication by HHV-6.92
Perhaps most interestingly, numerous investigators have demonstrated that HHV-6 can trans-activate the LTR promoter of HIV.74,75,81,94–104 Such transcriptional activation occurs in an NF-kB- or Spl-binding-site-dependent manner.75,95,97,102,104 Co-expression of HIV-1 tat transactivating protein produces enhanced transcriptional activation.75,81 Transactivation of the HIV LTR is accomplished by both HHV-6A and HHV-6B, although HHV-6B induction requires T cells to be stimulated, whereas induction by HHV-6A can occur in either stimulated or resting T cells.95
Loci of HHV-6A that are capable of HIV-1 LTR transactivation are depicted in Figure 52-1.19 These loci contain homologs of the CMV UL36-38 and CMV US22 gene families,95–100 a portion of the putative DNA polymerase accessory protein,103 putative IE genes,96 and sequences of unknown function.95,102
In Vivo Data
The consequence of HHV-6 on HIV infection in vivo has been inadequately evaluated. Of the few published clinical reports that have evaluated the potential interaction between these two viruses from epidemiologic and pathologic perspectives, conflicting results have emerged. In a study of the prevalence of HHV-6 seropositivity in a cohort of HIV-1-infected patients, the frequency of HHV-6 seropositivity was significantly lower in patients who had a slower decline in CD4 cells, compared to patients with rapidly declining CD4+ T-lymphocyte counts and to the general population.105 This finding suggests a role for HHV-6 co-infection in the progression of HIV-1 disease. HHV-6 DNA has been detected primarily in HIV-infected patients with high CD4+ T-lymphocyte counts in some studies75,106,107 and primarily in patients with low CD4+ T-lymphocyte counts in others.108 A small longitudinal study of HIV-infected patients has demonstrated that HHV-6 reactivation was followed by a temporary decrease in CD4+ T-lymphocyte counts and by a progressive, dramatic loss of CD4 cells during the following 18 months.109
HHV-6 DNA is present in the peripheral lymph nodes and spleens of recent HIV-1 seroconverters at a frequency and distribution similar to that of control tissues from individuals not infected with HIV.110 In patients with advanced HIV disease, however, HHV-6 can disseminate widely to visceral organs, including the lungs of patients who died of pneumonitis.111–113 AIDS patients have significantly higher HHV-6 viral loads in these visceral organs than do patients who are not infected with HIV.113 Furthermore, HIV-infected patients whose organs are co-infected with HHV-6 have higher concentrations of HIV proviral DNA in those organs, compared with HIV-infected persons who are not co-infected with HHV-6.39,75
HHV-6 DNA has been detected by PCR in the colonic mucosa of HIV-seropositive patients with diarrhea undergoing intestinal biopsy.114 A study involving serologic evaluation of Romanian children with nonprogressive HIV disease found that current or recent HHV-6 infection appeared to be associated with the development of pneumonitis,115 although other reports have not correlated HHV-6 with respiratory disorders in HIV-infected patients.116 HHV-6 has been correlated with pneumonitis in other immunocompetent and immunocompromised pediatric populations, however.117 Several reports have documented the presence of HHV-6 antigens and DNA in retinal lesions from patients with AIDS-associated retinitis, with CMV or HIV-1 usually being present concomitantly.118–122 In vitro studies have proven that HIV-1 and HHV-6 are capable of simultaneously infecting corneal epithelial cells.123 Several studies have suggested that HHV-6 can be detected infrequently from cerebrospinal fluid of HIV-infected persons, although its pathogenicity in such settings has been called into question and may relate to co-pathogens such as CMV.124–126
Although the studies cited above suggest that HHV-6 plays a role as cofactor in HIV disease progression, other reports have found no correlation between HHV-6 infection and the course of the HIV infection.127–132 Investigations in animal models have also failed to demonstrate a clinical consequence of simultaneous infection with HHV-6 and HIV.133
With limited exceptions,59,115 each of the in vivo studies to date involved only adult patients. Because HHV-6 establishes latency following primary infection during childhood, an inherent difficulty with clinical evaluations of the possible interaction(s) between HHV-6 and HIV-1 is distinguishing causality. That is, it is difficult to determine if HHV-6 reactivation results in progression of HIV-1 disease, or if progression of HIV-1 disease (with its corresponding decline in immunoregulatory function) results in reactivation of HHV-6. In an effort to assess the impact of primary HHV-6 acquisition on HIV viral dynamics and disease course, Thai and Japanese investigators evaluated 227 infants born to HIV-infected women. The cumulative infection rates of HHV-6 at 6 and 12 months of age were significantly lower in HIV-infected children (11% and 33%, respectively) than in uninfected children (28% and 78%, respectively; p < 0.001).59 Similar findings have been observed by one of the authors of this chapter in an analysis of specimens obtained from patients enrolled in the National Institutes of Health (NIH)-sponsored Women and Infants Transmission Study (WITS). HIV-infected infants without HHV-6 co-infection had lower rates of HIV disease progression at 12 months of life than did co-infected patients (42% vs 100%; p < 0.05), suggesting an association of HHV-6 infection and progression of HIV disease in children with vertically acquired HIV infection.59




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


