INTRODUCTION
SUMMARY
Classical Hodgkin lymphoma is derived by malignant transformation of a mature B cell at the germinal center stage of differentiation and is characterized pathologically by multinucleated Hodgkin and Reed-Sternberg cells embedded in a mixed infiltrate of nonneoplastic cells*. Hodgkin and Reed-Sternberg cells contain monoclonal immunoglobulin gene rearrangements, but have lost most of the B-cell–specific expression program. Multiple signaling pathways and transcription factors are deregulated in Hodgkin lymphoma and genetic lesions involving the JAK-STAT and nuclear factor-κB pathways are commonly identified. Epstein-Barr virus is an important environmental factor in the pathogenesis of Hodgkin lymphoma, and also leads to activation of the nuclear factor-κB pathway. The inflammatory microenvironment promotes survival and allows escape of Hodgkin and Reed-Sternberg cells from immune attack. Morphologic and immunophenotypic features distinguish the four subtypes of classical Hodgkin lymphoma (accounting for 95 percent of cases) from nodular lymphocyte predominance Hodgkin lymphoma (accounting for 5 percent of cases). Hodgkin lymphoma spreads in a predictable, contiguous manner and is classified into four stages, I to IV. Hodgkin lymphoma is treated with the intent to cure the disease in all stages, and long-term survival exceeds 85 percent. Doxorubicin-containing chemotherapy plays a major role in treatment of all stages of the disease whereas radiotherapy is used selectively because of concerns for late toxicities. 18-Fluorodeoxyglucose positron emission tomography is a valuable diagnostic test for assessment of disease extent and response to treatment. High-dose therapy and autologous transplantation are effective in patients who have relapsed, and several promising new biologic agents are available, including brentuximab vedotin (an anti-CD30 antibody–drug conjugate) and nivolumab (an anti-PD1 blocking antibody). Concerns regarding late treatment effects guide therapy and followup decisions in Hodgkin lymphoma, which disproportionately affects adolescents and young adults. Major treatment challenges include the maintenance of high cure rates with fewer short-term and long-term complications, biomarker identification of the small refractory subgroup, and integration of biologic therapies into treatment paradigms.
Acronyms and Abbreviations
ABVD, Adriamycin (doxorubicin), bleomycin, vinblastine, dacarbazine; AP1, activator protein 1; BCMA, B-cell maturation antigen; BEACOPP, bleomycin, etoposide, Adriamycin (doxorubicin), cyclophosphamide, vincristine, procarbazine, prednisone; BEAM, bischloroethylnitrosourea (carmustine), etoposide, Ara C (cytarabine), melphalan; CBV, cyclophosphamide, bischloroethylnitrosourea (carmustine), etoposide; cHL, classical Hodgkin lymphoma; COPP, cyclophosphamide, vincristine, procarbazine, prednisone; CT, computed tomography; DHAP, dexamethasone, cytarabine, cisplatin; EBV, Epstein-Barr virus; EBVP, epirubicin, bleomycin, vinblastine, prednisone; EORTC, European Organization for the Research and Treatment of Cancer; ERK, extracellular signal-regulated kinase; ESR, erythrocyte sedimentation rate; FDG, 18-fluorodeoxyglucose; FIL, Fondazione Italiana Linfomi; GELA, Groupe d’Etude des Lymphomes de l’Adulte; GHSG, German Hodgkin Study Group; GVD, gemcitabine, vinorelbine, liposomal doxorubicin; HLA, human leukocyte antigen; ICE, ifosfamide, carboplatin, etoposide; IL, interleukin; JAK, Janus kinase; LMP, latent membrane protein; LySA, Lymphoma Study Association; MOPP, mechlorethamine (nitrogen mustard), Oncovin (vincristine), procarbazine, prednisone; NF-κB, nuclear factor-κB; NLPHL, nodular lymphocyte-predominant Hodgkin lymphoma; OS, overall survival; PD1, cell death protein 1; PET, positron emission tomography; PI3K, phosphoinositide 3′-kinase; RANK, receptor activator of nuclear factor-κB; R-CHOP, rituximab, cyclophosphamide, hydroxydaunorubicin, vincristine (Oncovin), prednisone; STAT, signal transducer and activator of transcription; TACI, transmembrane activator, calcium modulator, and cyclophilin ligand interactor.
*Sandra J. Horning, MD, was the author of this chapter for the 8th edition of Williams Hematology and significant portions of that chapter have been retained.
DEFINITION AND HISTORY
Classical Hodgkin lymphoma (cHL) is a neoplasm of lymphoid tissue, in most cases derived from a germinal center B cell, defined by the presence of the malignant Hodgkin and Reed-Sternberg cells with a characteristic immunophenotype and appropriate cellular background. cHL accounts for 95 percent of cases and contains four histologic subtypes distinguished on the basis of microscopic appearance and relative proportions of Hodgkin and Reed-Sternberg cells, lymphocytes, and fibrosis (nodular sclerosis, mixed cellularity, lymphocyte-rich and lymphocyte-depleted; Table 97–1). Nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) represents the other major category, which is distinguished by Hodgkin and Reed-Sternberg variants termed lymphocytic and histiocytic cells that, unlike cHL, express typical B-lineage markers.
HISTORICAL ASPECTS
In his historic 1832 paper entitled On Some Morbid Appearances of the Absorbent Glands and Spleen, Thomas Hodgkin described the clinical histories and gross postmortem findings of seven cases of the disease that was later to bear his name.1 In 1856, Samuel Wilks independently described 10 cases of “a peculiar enlargement of the lymphatic glands frequently associated with disease of the spleen,” including four of Hodgkin’s original cases.2 Upon discovering Hodgkin’s original report, he used the appellation “Hodgkin’s Disease” in a subsequent series of 15 cases published in 1865.2 Thirteen years after Hodgkin’s original paper, the first cases of leukemia were described. Cases in which the neoplastic cells remained confined to the lymphatic system were described by Dreschfield (1892)3 and Kundrat (1893)4; Kundrat gave the name lymphosarcoma to these cases. The description of additional members of the lymphoma–leukemia complex continued up to the present time.
Carl Sternberg (1898)5 and Dorothy Reed (1902)6 are credited with the first definitive and thorough descriptions of the pathology of cHL, although a number of investigators from England, Germany, and France had previously recognized the characteristic multinucleated giant cells. In 1926, Fox examined microscopic sections from the gross specimens preserved in the Gordon Museum of Guy’s Hospital in London of three of Hodgkin’s original cases.7 It is remarkable that the preserved microanatomy allowed him to confirm the histopathologic diagnosis in two of these cases. Jackson and Parker made the first serious effort at the histopathologic classification of cHL, correlating their findings with prognosis.8 A second advance was made in 1966 when Lukes, Butler, and Hicks proposed a classification that related well to clinical presentation and course.9 Their proposal was slightly modified into the Rye classification, in which four histopathologic subtypes were described: lymphocyte-predominant, nodular sclerosis, mixed cellularity, and lymphocyte-depleted. In the World Health Organization Classification of Lymphoid Neoplasms, the NLPHL subtype is clearly distinguished from cHL.10 The “lymphocyte-rich” subtype of cHL was introduced in 1999.
Peters described a clinical staging system in 1950, emphasizing the diagnostic evaluation of the anatomic extent of disease.11 In 1952, Kinmouth introduced lower-extremity lymphangiography that allowed roentgenologic visualization of the pelvic and retroperitoneal lymph nodes and was found to be far more sensitive than palpation or other radiographic methods.12 The frequency of unsuspected splenic involvement was revealed in a group of 65 patients subjected to laparotomy and splenectomy with biopsy of splenic hilar, paraaortic and mesenteric nodes, and liver at Stanford University.13 These diagnostic procedures led to improved understanding of the mode of dissemination of the disease and correlated well with prognosis, culminating in the modern concepts of staging codified at the Rye, New York, conference in 1965,14 and further refined at the Workshop on the Staging of Hodgkin’s Disease in Ann Arbor, Michigan, in 1971.15
Pusey (1902)16 and Senn (1903)17 were the first to report dramatic regressions of lymphadenopathy with exposure to X-rays, discovered by Roentgen in 1896. Based upon the nearly inevitable recurrence in untreated areas, Gilbert proposed the systematic treatment of both involved and uninvolved areas in 1939.18 Peters (1950) is credited for the first demonstration of the curative potential of radiotherapy in her classic paper.11 The development of megavoltage radiotherapy (doses >4000 cGy), as reported by Kaplan in 1962,19 permitted the delivery of tumoricidal doses to virtually all lymphoid regions in the body within acceptable limits of normal tissue tolerance.
The chemotherapy of cHL originated as a byproduct of the wartime work on the mustard gases.20,21 Following the initial work with the nitrogen mustards, antimetabolites were synthesized and a number of alkaloids and antibiotics extracted from various plant, fungus, and microbial sources became available for clinical use. DeVita and colleagues introduced the first highly effective combination chemotherapy, MOPP (mechlorethamine [nitrogen mustard], vincristine [Oncovin], procarbazine, and prednisone), based on experimental studies indicating the desirability of combining agents with non-overlapping toxicities.22 Combination chemotherapy extended the curative potential for cHL to advanced disease. The ABVD (doxorubicin [Adriamycin], bleomycin, vinblastine, dacarbazine) regimen introduced by Bonadonna and colleagues represented another major advance.23 Based on a more favorable safety profile and greater efficacy, ABVD replaced MOPP.
EPIDEMIOLOGY
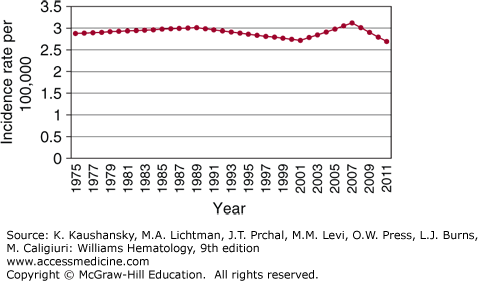
The estimated incidence of Hodgkin lymphoma in the United States was 9190 cases in 2014, with equal incidences in Americans of European and African descent (2.9/100,000).24 The disease has a median age of onset of 38 years with bimodal incidence peaks at ages 15 to 34 and older than age 60 years (Fig. 97–1).25 The second incidence peak is smaller in Americans of European descent whereas it is more prominent in Americans of Hispanic descent.24 Except for Americans of Asian descent, for whom the incidence has increased by 5.2 percent per year, the incidence of Hodgkin lymphoma has been stable in the United States from 1975 to 2011 (Fig. 97–2). The nodular sclerosis subtype predominates in young adults, whereas the mixed cellularity subtype is more common in the pediatric population and at older ages. There is a male predominance at all ages (~1.4:1).
Figure 97–1
The graph depicts the incidence of Hodgkin lymphoma as a function of age among American males and females, 2000 to 2011. (Data from the Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) Research Data (1973-2011), National Cancer Institute, DCCPS, Surveillance Research Program, Surveillance Systems Branch, released April 2014, based on the November 2013 submission; 2014.)
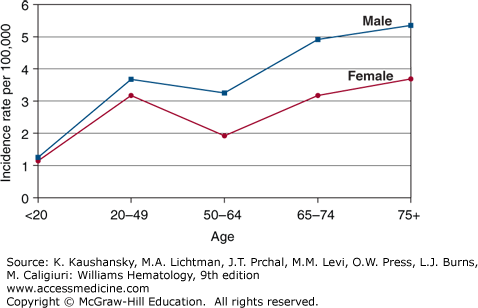
Figure 97–2.
Incidence of Hodgkin lymphoma by calendar year. (Data from the Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) Research Data (1973-2011), National Cancer Institute, DCCPS, Surveillance Research Program, Surveillance Systems Branch, released April 2014, based on the November 2013 submission; 2014.)
Early studies associated an increased risk of Hodgkin lymphoma in the young adult population with high socioeconomic status.26 Living in a rental home, sharing a bedroom, and attending daycare or nursery school and early parity in women have been associated with reduced risk. The relationship of incidence to neighborhood socioeconomic status was demonstrated in California for younger but not for older patients.25 Although associations with occupational exposure to pesticides and lifestyle factors such as cigarette smoking have been reported, the aggregate data do not indicate consistent causal relationships with exogenous chemicals or toxins. A personal or family history of an autoimmune disorder, particularly sarcoidosis, has been associated with an increased risk of Hodgkin lymphoma.27 Shared etiologic factors with multiple sclerosis have been suggested but these are thought to be of minor importance.
Geographic patterns vary for the three major age groups: the incidence of Hodgkin lymphoma is greater in childhood in less-developed countries, whereas the incidence peaks in young adulthood and is associated with more favorable histologic subtypes in developed countries.28 Presence of the Epstein-Barr virus (EBV) in Hodgkin and Reed-Sternberg cells is more common in less-developed countries and in pediatric and older adult cases. The worldwide incidence of the disease is much lower in the Asian population, whether residing in the Far East or in the United States, although the reported rate in Vancouver, Canada, among immigrants of Chinese descent was higher than among Chinese residing in Hong Kong.29,30 Together these data suggest a complex interaction among possible socioeconomic, environmental, immunologic, genetic, and infectious factors in the incidence of Hodgkin lymphoma.
POSSIBLE INFECTIOUS ETIOLOGY
Demographic features have long supported a “hygiene hypothesis” postulating that one or more subtypes of cHL represent delayed exposure to an infectious agent. In 1966, MacMahon proposed that the first age peak in young adults was infectious in nature, whereas the second peak resulted from causes similar to other lymphomas.26 As noted above, socioeconomic status correlates with the first, but not the second, peak.25 Several reports of clustering of cHL at the time of diagnosis suggested the possibility of infectious transmission.31 The weaknesses of the retrospective methodology in these studies have been critically assessed, and further statistical analyses indicate that these likely occurred by chance alone.
A threefold increased risk of cHL in young adults is conferred by a prior history of serologically confirmed infectious mononucleosis. In addition, elevations in titers of EBV, the etiologic agent of infectious mononucleosis, have been reported in patients with cHL.32,33 A large population study showed that people who developed the disease had abnormally high titers of EBV viral capsid antigen and early antigen in prediagnostic sera.34 In two subsequent reports, a significantly increased risk of cHL after serologically verified infectious mononucleosis and limited to EBV-positive cases was reported in young adults.35,36 The median incubation time was approximately 4.1 years.
EBV genomes have been detected in 30 to 50 percent of cHL tissues in developed countries, and EBV-associated cases are more common in cases with mixed cellularity histology, Hispanic ethnicity, and patients older than the age of 60 years.37,38,39 Several studies report a high incidence of EBV association, 85 to 100 percent, in pediatric cHL in which geographic, ethnic, and racial factors have been implicated in the association.37,38,39 The incidence of cHL is 10 to 20 times higher in patients with HIV infection than in the general population, and such cases typically have detectable EBV within Hodgkin and Reed-Sternberg cells.40 In contrast to non-Hodgkin lymphoma, the incidence of cHL in the HIV-infected population has increased despite less-severe immunosuppression in the era of highly active antiretroviral therapy.41,42
GENETIC BASIS
Genetic susceptibility and familial aggregation appear to play a role in the incidence of cHL. The increased risk of the disease among identical, but not fraternal, twins provides the strongest evidence for a genetic association.43 cHL-prone families, with or without other forms of cancer, have been described in the literature and it is estimated that 4.5 percent of cases are familial.44,45,46 The standard incidence ratio for age-specific familial risk from the Swedish Cancer Registry was higher for Hodgkin lymphoma (4.8) than any other neoplasm.47 The relative risk for familial disease is stronger in individuals older than 40 years of age, males, and siblings, and a shared risk with chronic lymphocytic leukemia and non-Hodgkin lymphoma has been described.46 An increased incidence in same sex siblings (eight- to 12-fold) versus opposite-sex siblings (1.3- to 1.4-fold) detected in the Swedish registry is consistent with older data and has been interpreted to be supportive of an environmental influence or a pseudoautosomal susceptibility gene located on a sex chromosome.48,49,50
Immunoregulatory genes within or near the major histocompatibility complex that may govern susceptibility to viral infections have been postulated to influence susceptibility to cHL, an hypothesis that is supported by the demonstration of lifelong, depressed cellular immunity in cHL patients and their healthy relatives.51 Several groups described specific human leukocyte antigen (HLA) susceptibility or resistance regions, but these data have been relatively weak and sometimes inconsistent.52
ETIOLOGY AND PATHOGENESIS
The histologic diagnosis of cHL is based on the recognition of the Reed-Sternberg cell in an appropriate cellular background. The classic Reed-Sternberg cell has a bilobed nucleus with prominent eosinophilic nucleoli separated by a clear space from the thickened nuclear membrane (Fig. 97–3; see also Chap. 96, Fig. 96–35). Mononuclear variants (Hodgkin cells) have similar nuclear characteristics and may represent Reed-Sternberg cells cut in a plane that shows only one lobe of the nucleus. Reed-Sternberg cells are not pathognomonic for cHL; they may be seen in reactive and other neoplastic conditions. Study of the Reed-Sternberg cell has been complicated by the fact that the neoplastic cells are sparsely interspersed among a reactive, mixed, nonclonal population of lymphocytes, eosinophils, histiocytes, plasma cells, and neutrophils. Difficulty characterizing the neoplastic cells, which account for only 1 to 2 percent of the cellular composition, led to controversy regarding the etiology and pathogenesis of cHL for more than 150 years. Molecular analyses of single cells obtained by microdissection led to the discovery that cHL and NLPHL are both clonal disorders derived from germinal center B cells, in most cases.53 The need to survive negative selection in the germinal center, the determination of genetic alterations and constitutive activity of key signaling pathways, and the involvement of EBV in a subset of cases have led to hypotheses concerning the malignant transformation events leading to the formation of Hodgkin/Reed-Sternberg cells. Application of additional genomic technology promises to clarify the molecular changes underlying malignant transformation and cellular proliferation.
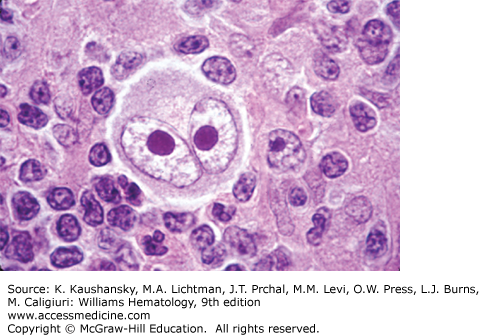
Reed-Sternberg cells and their mononuclear variants demonstrate inconsistent lineage-specific antigen expression that is unlike any other cell of the hematopoietic system. The origin of these cells was eventually determined through isolation of single cells by micromanipulation of histologic sections and analysis for immunoglobulin variable gene rearrangements.53,54 Nearly all Hodgkin and Reed-Sternberg cells have rearranged and somatically mutated immunoglobulin VH genes, indicating a germinal center or postgerminal center origin of classic Hodgkin and Reed-Sternberg cells.55,56,57 Extrapolating from the fact that a subset of these cells carries crippling mutations, it is possible that Hodgkin and Reed-Sternberg cells originate from a preapoptotic germinal center B cell with unfavorable mutations that has escaped negative selection. Rare cases of cHL with a clonal T-cell receptor gene rearrangement have been observed.58 In contrast, single-cell analyses of NLPHL demonstrated clonal immunoglobulin gene rearrangements with ongoing mutations, an intraclonal diversity consistent with a germinal center origin of lymphocyte and histiocytic cells.59,60,61
Hodgkin and Reed-Sternberg cells show a global loss of their B-cell phenotype, retaining only B-cell features associated with their interaction with T cells and their antigen-presenting function.62 Furthermore, Hodgkin and Reed-Sternberg cells express markers of other lineages, including T cells, dendritic cells, cytotoxic cells, and myeloid cells. The lack of expression of numerous B-cell genes is the result of loss of transcription factor expression (OCT2, BOB1, PU.1) and epigenetic silencing.63,64,65 The main B-cell lineage commitment factor, PAX5, is typically expressed, but its target genes are downregulated.66,67 Reduced expression of target genes likely reflects the fact that B-cell genes are regulated by coordinated action of multiple transcription factors.
The heterogeneity of expression of myeloid, T-cell, dendritic cell, and other genes by Hodgkin and Reed-Sternberg cells is the result of many factors. Early B-cell factor 1 levels are low, de-repressing the expression of T-cell and myeloid genes and lowering transcription of B-cell–specific genes.68 Notch 1, which plays a key role in promoting T-cell differentiation and inhibiting B-cell development, is expressed in Hodgkin and Reed-Sternberg cells.69 Notch 1 also contributes to the expression of GATA2, a transcription factor required for proliferation and survival of hematopoietic stem cells.70 The hematopoietic stem cell regulator polycomb G proteins are also expressed by Hodgkin and Reed-Sternberg cells and are thought to contribute to the expression of markers of different hematopoietic lineages.71 The signal transducer and activation of transcription factors (STAT) 5A and STAT5B are implicated in Hodgkin and Reed-Sternberg reprogramming as they upregulate CD30 and downregulate B-cell–receptor expression.72 Together, these factors cause a global loss of the B-cell phenotype and aberrant expression of genes of other cell lineages.
Because Hodgkin and Reed-Sternberg cells lack expression of functional B-cell surface receptors, rescue from apoptosis is probably an important mechanism of survival.53,73 The most prevalent genetic lesions in Hodgkin and Reed-Sternberg cells involve two signaling pathways: Janus kinase (JAK)-STAT and nuclear factor-κB (NF-κB). Hodgkin and Reed-Sternberg cells have frequent gains in JAK2 and inactivation of the negative regulator of JAK-STAT signaling, suppressor of cytokine signaling 1, resulting in enhanced cytokine signaling.74,75 Genetic alterations in NF-κB include gains and amplifications of the NF-κB transcription factor REL in about half the cases of cHL.76 Somatic mutations of the gene encoding the inhibitor of NF-κB (IκBα) occur in approximately 20 percent of cases.77,78 Inactivating mutations and deletions of the gene encoding A20, a negative regulator of NF-κB, have been found in approximately 40 percent of cases, nearly all of which were EBV-negative.79
Autocrine and paracrine signaling events also contribute to constitutive activation of the JAK-STAT pathway and NF-κB transcription.72 STAT factors are activated by autocrine means through expression of interleukins (ILs) 13 and 21 and their receptors by Hodgkin and Reed-Sternberg cells and augmented by NF-κB activity.80,81,82 Receptor tyrosine kinases expressed in these cells may also contribute to STAT activation. The tumor necrosis factor receptor family, which includes CD30, CD40, transmembrane activator, calcium modulator, and cyclophilin ligand interactor (TACI), B-cell maturation antigen (BCMA), and receptor activator of NF-κB (RANK), is involved in NF-κB signaling through interactions with the cHL microenvironment or in an autocrine fashion.83,84
Multiple receptor tyrosine kinases are aberrantly expressed in Hodgkin and Reed-Sternberg cells, including platelet-derived growth factor receptor-α. In addition, deregulated and constitutive activation of the phosphoinositide 3′-kinase (PI3K)-AKT and extracellular signal-regulated kinase (ERK) pathways are implicated in Hodgkin and Reed-Sternberg cells. The activator protein 1 (AP1) transcription factors also appear to play a role, inducing target genes such as galectin 1 and CD30 in Hodgkin and Reed-Sternberg cells.
Several factors point to the pathogenetic role of EBV in approximately 40 percent of classical cHL. The viral proteins latent membrane protein 1 (LMP1) and latent membrane protein 2 (LMP2), in particular, appear to have hijacked signaling pathways to promote the survival of EBV-infected Hodgkin and Reed-Sternberg cells. LMP1 induces constitutive NF-κB signaling by mimicking the CD40 receptor and can activate JAK-STAT, PI3K, and AP1 signaling. LMP2 functions as a surrogate for the B-cell receptor. The role of EBV in the pathogenesis of cHL also is supported by the findings that (1) there is an inverse relationship between expression of multiple receptor tyrosine kinases and EBV expression, (2) there is an ability of EBV to rescue crippled germinal center B cells in the laboratory, (3) mutations preventing any B-cell receptor expression are in EBV-positive Hodgkin and Reed-Sternberg cells, and (4) there is an inverse relationship between mutations reducing the expression of the NF-κB regulator A20 and EBV-positive Hodgkin and Reed-Sternberg cells.
Overall, genetic alterations involving the JAK-STAT and NF-κB signaling pathways and further activation via autocrine or paracrine mechanisms interact to support the growth and survival of cHL cells. In the EBV-positive subset of patients, viral genes can provide the pathogenetic function of genetic lesions found in EBV-negative cases.
The survival of Hodgkin and Reed-Sternberg cells appear to be dependent on their microenvironment, which represents 95 to 99 percent of the cellular composition of the tumor. Hodgkin and Reed-Sternberg cells attract T cells, B cells, neutrophils, plasma cells, eosinophils and mast cells by secreting chemokines (Fig. 97–4). For instance, CCL5, CCL17, and CCL22 attract T-helper 2 and T-regulatory cells. Other chemokines attract eosinophils and mast cells, and IL-8 attracts neutrophils. These chemokines may also have direct effects on Hodgkin and Reed-Sternberg cells. T cells represent the largest and probably most important population. CD4+ T cells trigger CD40 signaling and CD4+ T-regulatory cells have potent immunosuppressive activity against infiltrating cytotoxic T cells. Other interactions include activation of TACI and BCMA through their ligand production by neutrophils and activation of CD30 through CD30 ligand-expressing mast cells and eosinophils. Connective tissue cells and their products can be involved in complex interactions, such as the stimulation of fibroblasts via factors expressed by Hodgkin and Reed-Sternberg cells and the consequent secretion by these fibroblasts of eotaxin and CCL5, which attract eosinophils and T-regulatory cells to the cHL microenvironment.
Figure 97–4.
The Reed-Sternberg cell and its environment. In a network of highly complex interactions, Reed-Sternberg cells elaborate chemokines that attract a variety of cells, which cascade their influence on the microenvironment. Reed-Sternberg cells express ligands that play a role in autocrine and paracrine interactions and also produce a number of immunosuppressive factors (e.g., galectin 1) that directly contribute to a protumor, humoral T-helper 2 environment (see “Role of the Microenvironment” for further details).
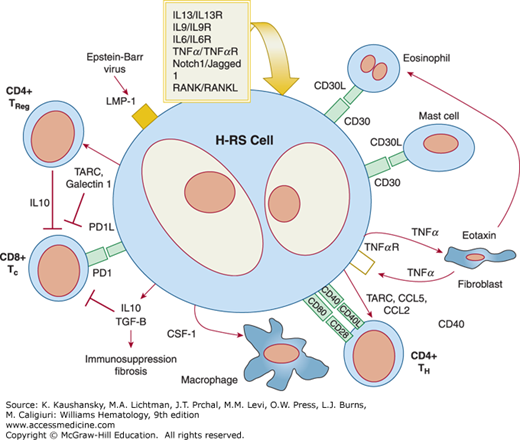
The differentiation of CD4+ T cells to T-regulatory cells and the immunosuppressive features of the cHL microenvironment have received much attention. A hallmark of these changes is the shift from an antitumor, cytotoxic T-helper 1 response to a protumor, humoral T-helper 2 response. Hodgkin and Reed-Sternberg cells produce a number of immunosuppressive factors such as IL-10, transforming growth factor-β, galectin 1, and prostaglandin E2. Hodgkin and Reed-Sternberg cells express programmed cell death protein 1 (PD1) ligand that binds and inhibits T-cell cytotoxic function.
CLINICAL FEATURES
Constitutional symptoms accompany the diagnosis of cHL in approximately 30 percent of cases. Fever in excess of 38°C, drenching night sweats, and weight loss exceeding 10 percent of baseline body weight during the 6 months preceding diagnosis are designated as symptomatic “B” disease. Fevers are usually of low grade and irregular. Rarely, a cyclic pattern of high fevers for 1 to 2 weeks alternating with afebrile periods of similar duration, known as Pel-Ebstein fever, is present at diagnosis and is virtually diagnostic of the disease.85,86 Generalized pruritus, often accompanied by marked excoriation, may be present at diagnosis; but does not confer prognostic significance. Pain in involved lymph nodes immediately after the ingestion of alcohol is a curious complaint that occurs in fewer than 10 percent of patients but is nearly specific to cHL.87 The etiology of these symptoms has been the subject of speculation, but remains largely unexplained. Patients with extensive intrathoracic disease may present with cough, chest pain, dyspnea, and, rarely, hemoptysis. Infrequently, patients present with bone pain, including the constellation of back pain accompanied by signs and symptoms of spinal cord compression.
Detection of an unusual mass or swelling in the superficial, supradiaphragmatic lymph nodes (60 to 70 percent cervical and supraclavicular, 15 to 20 percent axillary) is the most common presentation of cHL. Only 15 to 20 percent of patients have subdiaphragmatic disease at presentation.88 Lymphadenopathy is usually nontender and has a “rubbery” consistency. By inspection, a diffuse, puffy swelling rather than a discrete mass may be apparent in the supraclavicular, infraclavicular, or anterior chest wall regions. Infrequently, compression of the superior vena cava will result in facial swelling and engorgement of the veins in the neck and upper chest. Auscultation of the chest may reveal a pleural effusion. Rarely, a significant pericardial effusion is present at diagnosis. Palpation of the abdomen may reveal intraabdominal masses or hepatosplenomegaly, although physical examination is relatively insensitive for detection of these abnormalities.
A number of rare paraneoplastic syndromes have been described in cHL at the time of diagnosis. These include “vanishing bile duct syndrome” and idiopathic cholangitis with clinical jaundice, the nephrotic syndrome with anasarca, autoimmune hematologic disorders (e.g., immune thrombocytopenia or hemolytic anemia), and neurologic signs and symptoms.89,90,91 Although parenchymal involvement of the central nervous system or meningeal involvement is rare in cHL, paraneoplastic syndromes include subacute cerebellar degeneration, myelopathy, progressive multifocal encephalopathy, and limbic encephalitis.91,92
Intrathoracic disease is present at diagnosis in two-thirds of patients. Mediastinal adenopathy is common in cHL, particularly in young women with the nodular sclerosis subtype.93 Hilar adenopathy, pulmonary parenchymal involvement, pleural effusions, pericardial effusions, and chest wall masses may be appreciated by chest computed tomography (CT); these are more common in the presence of extensive mediastinal disease. CT of the abdomen and pelvis is routinely employed in the diagnostic evaluation of cHL. Although technologic advances have greatly increased the resolution of this technique and the subsequent detection of celiac, portal, splenic hilar, and mesenteric lymph nodes, the correlation with histologic involvement of the spleen, historically determined by laparotomy staging, has been disappointing.
Whole-body 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) has become standard in the staging of cHL (Fig. 97–5).94,95 FDG-PET correlates well with CT evaluation and may demonstrate additional areas of disease, although this information uncommonly results in changes in stage or choice of initial therapy.95,96 FDG-PET, however, is more sensitive to bone and hepatic disease and a diffuse increase in signal can be seen at diagnosis in patients with neutrophilia. FDG-PET imaging is superior to CT scanning in distinguishing active residual disease (increased glucose metabolism) from inactive residual tissue, a major problem in assessing remission status after treatment, and has been incorporated in formal revised response guidelines.94,95 False-positive FDG-PET scans can be seen in the marrow during or at the end of treatment as a consequence of the chemotherapy effect or use of hematopoietic colony-stimulating factors. In followup, false-positive studies may be caused by thymic hyperplasia, granulomatous disease, or infectious disorders. In addition to evaluation of residual masses, FDG-PET has been incorporated in early response monitoring for risk stratification and, in clinical trials, to alter therapy.96,97,98,99,100,101,102 The predictive accuracy of FDG-PET is dependent on expertise of the imaging staff and clinical correlation. In most situations, but particularly in FDG-PET, avid anatomic sites that were previously uninvolved or those without concomitant abnormality on CT scan, usually require tissue biopsy confirmation. Combined CT and FDG-PET technology is now standard for staging and posttreatment response evaluation and has resulted in improved anatomic definition of sites with increased signal.94,95,96
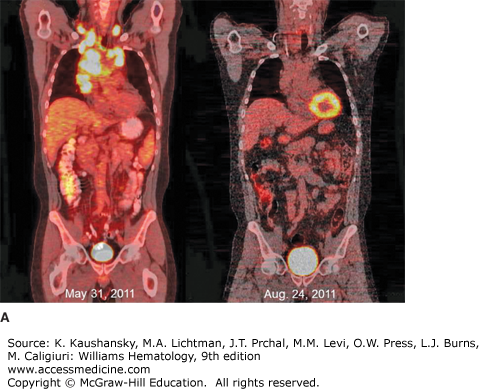
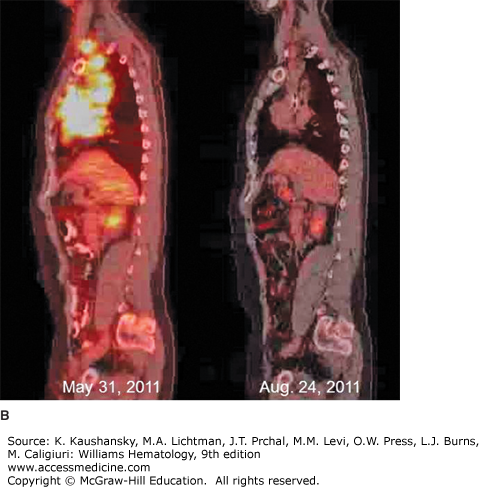
Figure 97–5.
18Fluorodeoxyglucose positron emission tomography/computed tomography (FDG-PET/CT) imaging of classical Hodgkin lymphoma. A. Coronal views of whole-body FDG-PET/CT imaging performed before (May 31, 2011) and after (August 24, 2011) therapy for stage IIB classical Hodgkin lymphoma with Adriamycin, bleomycin, vinblastine, and dacarbazine (ABVD) chemotherapy and involved field radiotherapy. At the time of diagnosis, hypermetabolic areas of avid-FDG uptake were evident in bilateral cervical, supraclavicular, mediastinal, and hilar lymph nodes. Physiologic FDG uptake was visible in the colon and bladder. After completion of therapy, there was no abnormal hypermetabolic activity in any of the original sites of disease, although physiologic FDG activity was seen in the cardiac blood cardiac blood pool. B. Sagittal views of the same patient before (May 31, 2011) and after (August 24, 2011) therapy, showing resolution of hypermetabolism in all involved nodal sites. The patient has remained in complete remission since finishing therapy.
CLINICAL AND PATHOLOGIC CORRELATION
There is a strong correlation between age at onset, the anatomic extent of disease and histologic subtype of Hodgkin lymphoma. Approximately 5 to 10 percent of patients present with NLPHL, which is considered to be a distinct subtype, discrete from cHL.103,104 Progressive transformation of germinal centers may precede or follow NLPHL in other sites. The cellular composition is predominantly benign B lymphocytes with or without histiocytes. The characteristic multilobated, CD20+ lymphocyte and histiocytic cells are relatively abundant (see Chap. 96, Fig. 96–40). Patients most commonly present with stage I disease (70 percent) in peripheral lymph node sites, particularly in the axillae, and there is a 3 to 4:1 male predominance.103,104 NLPHL may be associated with diffuse large B-cell non-Hodgkin lymphoma as a composite tumor or a large cell lymphoma may develop at a later date.105,106 The large cell variant, T-cell–rich B-cell lymphoma, may be difficult to distinguish from NLPHL and may occur concurrently or subsequently (Chap. 98).107
cHL is characterized by Hodgkin and Reed-Sternberg cells that express CD30 and CD15+ but usually not typical B-cell surface markers such as CD20 or CD79B. The nodular sclerosis subtype constitutes 40 to 70 percent of cHL and is distinguished by its distinctive clinical and histologic features. This subtype typically involves lower cervical, supraclavicular, and mediastinal lymph nodes in adolescents and young adults, particularly females. Approximately 70 percent of patients with the nodular sclerosis subtype present with limited stage disease. A characteristic histologic feature is the lacunar cell, a Reed-Sternberg variant that results from retraction of the cytoplasm of Hodgkin and Reed-Sternberg cells during formalin fixation (see Chap. 96, Fig. 96–38). Another typical feature is the thickened capsule and fibrous bands that divide the lymphoid tissue into cellular nodules.
Mixed cellularity cHL involves both pediatric and older age groups and is more commonly associated with advanced stage disease, constitutional symptoms, and immunodeficiency. Approximately 30 to 50 percent of patients present with this histology. Classic Hodgkin and Reed-Sternberg cells are easily found amid a cellular background composed of lymphocytes, eosinophils, plasma cells, and histiocytes (see Chap. 96, Fig. 96–39). A worse prognosis has been reported for this subtype.108
Lymphocyte-depleted cHL has two morphologic variants: reticular and diffuse fibrosis. The reticular variant contains abundant pleomorphic neoplastic cells whereas the diffuse fibrosis variant has a prominent fibroblastic proliferation with few normal lymphocytes. Hodgkin and Reed-Sternberg cells are sparse. The lymphocyte-depleted subtype presents in older patients with symptomatic, extensive disease, and may be associated with fever of unknown origin, jaundice, hepatosplenomegaly, or pancytopenia. Peripheral and mediastinal adenopathy is much less common than in other subtypes. Cases of cHL developing in the setting of HIV typically exhibit the lymphocyte-depleted morphology.
The lymphocyte-rich subtype of cHL was introduced by the World Health Organization classification in 1999 (see Table 97–1) following an expert pathology review of cases of NLPHL.109 The two subtypes differ subtly on morphologic grounds, but the major difference is that the Hodgkin and Reed-Sternberg cells in lymphocyte-rich cHL exhibit the classic CD30+ CD20− immunophenotype. The presenting features are very similar although patients with the lymphocyte-rich subtype tend to be older compared with NLPHL patients.109 A higher rate of multiple relapses and a more favorable prognosis upon relapse is characteristic of NLPHL.
ANATOMIC DISTRIBUTION OF DISEASE
In approximately 70 percent of patients, cHL presents in the cervical nodes; in 12 percent, in the axillary nodes; and in 9 percent, in the inguinal nodes.110 A small minority of patients presents exclusively with subdiaphragmatic disease. In an historical series of 285 consecutive, unselected, and untreated patients evaluated at Stanford University, involvement of abdominal lymph nodes and the spleen was documented in 272 upon laparotomy, a surgical diagnostic procedure in which the intraabdominal and pelvic lymph nodes are biopsied, the spleen is removed and examined pathologically in thin slices, the liver is biopsied by needle and wedge technique, and the marrow is biopsied. The frequency of splenic involvement at laparotomy in untreated patients averaged 37 percent in 17 published series.110 Involvement of the spleen was strongly dependent on histologic subtype: it was involved in 60 percent of mixed cellularity and lymphocyte-depleted cases compared with 34 percent of nodular lymphocyte-predominant and nodular sclerosis cases. Hepatic and marrow disease were invariably associated with splenic involvement.
Two different theories, the “contiguity” theory of Kaplan and Rosenberg111 and the “susceptibility” theory of Smithers,112 have been proposed for the mode of spread of cHL. In support of the former, most cases of cHL appear to spread via lymphatic channels to contiguous lymphatic structures in a predictable, nonrandom pattern. Controversy has surrounded the mode of spread to the spleen, which lacks afferent lymphatics. When four or more lymph node regions are involved, spread by hematogenous distribution appears likely. Disseminated disease is more common in mixed cellularity and lymphocyte-depleted cases, consistent with the presence of reported vascular invasion.113 Whereas vascular invasion is controversial, it is more common in the spleen than in lymph nodes and connotes a poor prognosis.114
STAGING
Optimal management of Hodgkin lymphoma relies on knowledge of the extent of disease dissemination, which is determined by performing a careful physical examination, supplemented by laboratory tests and radiographic imaging studies (Chap. 95). This process, known as “staging,” has been refined by a series of international working groups. The current 2014 Lugano staging system94 is a refinement of the historical Ann Arbor classification15 and assigns patients to one of four stages as indicated in Table 97–2. The classification for Hodgkin lymphoma is further refined by designating the presence or absence of constitutional “B” symptoms (fever >38°C, weight loss >10 percent of body weight, or drenching night sweats). Extranodal disease, representing extracapsular extension of lymph node disease that could be incorporated in a standard radiotherapy field, is distinguished from disseminated, stage IV disease. The correlation of this staging classification system with prognosis was extensively verified when radiotherapy served as the principal treatment for all but stage IV disease. Recommended staging procedures for untreated patients have evolved with changes in therapy. Exploratory laparotomy and splenectomy, which historically advanced about one-third of clinical stages I and II patients to pathologic stages III and IV, is no longer performed. CT of the chest, abdomen, and pelvis and FDG-PET imaging provide sensitive delineation of involved sites, and the use of chemotherapy in all stages of disease has reduced the critical nature of detecting subclinical disease. Marrow involvement occurs in approximately 12 percent of new patients and is more common in patients of older age, advanced stage, less-favorable histology, or those with constitutional symptoms or immunodeficiency. Because the marrow is almost never involved in young, asymptomatic patients with favorable clinical stage I or II presentations, marrow biopsy may be omitted in staging such patients.
Stage
|
Modifying Features
|
LABORATORY FEATURES
There are no diagnostic laboratory features of cHL. A complete blood count may reveal granulocytosis, eosinophilia, lymphocytopenia, thrombocytosis, or anemia. Anemia is usually the result of “chronic disease,” but rarely may be caused by hemolysis associated with a positive direct antiglobulin (Coombs) test. Thrombocytopenia may occur as a result of marrow involvement, hypersplenism or an immune mechanism. Immune neutropenia can occur in cHL. Cytopenias are particularly common in advanced-stage disease and the lymphocyte-depleted subtype. Elevation of the erythrocyte sedimentation rate (ESR) is most common in advanced disease and correlates with constitutional symptoms.115,116 The degree of sedimentation rate elevation correlates with prognosis, particularly in limited-stage disease.117 Although nonspecific, it may be a useful harbinger of recurrent disease if serially monitored. Serum lactate dehydrogenase levels are elevated in 35 percent of patients at diagnosis.118,119 The alkaline phosphatase may be elevated in cHL, nonspecifically in limited disease, or in association with involvement of liver, bone, or marrow in advanced disease. Hypercalcemia is unusual in cHL and when present may be secondary to synthesis of increased levels of 1,25-dihydroxyvitamin D by Hodgkin lymphoma cells.120 A variety of other abnormalities have been reported, including hypoglycemia121,122 resulting from autoantibodies to insulin receptors and hyponatremia resulting from inappropriate secretion of antidiuretic hormone.123
Anemia, granulocytosis, lymphopenia, and low serum albumin constitute four of seven adverse prognostic factors identified in advanced Hodgkin lymphoma by an international consortium.124 Similar to the non-Hodgkin lymphomas, serum β2-microglobulin levels correlate with tumor burden and prognosis in Hodgkin lymphoma.125 Serum levels of cytokines including soluble CD30, IL-6, IL-10, and the IL-2 receptor, have been reported to correlate with constitutional symptoms and advanced disease.126,127,128,129 Examination of pleural fluid in Hodgkin lymphoma may reveal transudative, exudative or chylous characteristics. Because cytology rarely yields diagnostic Hodgkin and Reed-Sternberg cells, the etiology is most often considered to be one of central lymphatic obstruction. Laboratory abnormalities, including abnormal liver function tests associated with marked enlargement of porta hepatis nodes and biliary obstruction or intrahepatic cholestasis, may be prominent in rare presentations of Hodgkin lymphoma.130 Similarly, the nephrotic syndrome, characterized by edema, azotemia, hypoalbuminemia, and hyperlipidemia, may rarely be present at the time of diagnosis of Hodgkin lymphoma.131
DIFFERENTIAL DIAGNOSIS
Clinically enlarged lymph nodes may be associated with a variety of infectious, inflammatory, autoimmune, and neoplastic disorders. Biopsy of unexplained, persistent, or recurrent adenopathy should be reviewed by an experienced hematopathologist. Distinction of cHL132 from primary mediastinal B-cell lymphoma may be difficult based on both clinical and histologic features. Gene expression profiling studies suggest that these disorders are closely related pathogenetically.132,133,134,135 Mixed cellularity Hodgkin lymphoma may demonstrate varied cellular and stromal compositions and must be distinguished from peripheral T-cell lymphoma and T-cell–rich, B-cell lymphoma, which can also be difficult to distinguish from NLPHL.136 Immune markers of Hodgkin and Reed-Sternberg cells, such as CD30, CD20, and CD15, are invaluable for differential diagnosis (see Chap. 96, Figs. 96–36 and 96–41). Nonneoplastic conditions that simulate Hodgkin lymphoma include viral infections, particularly infectious mononucleosis. Depleted nodes of any histology, including the depleted phase of lymph nodes from HIV-infected patients, may resemble the diffuse fibrosis variant of lymphocyte-depleted Hodgkin lymphoma. Histologic assessment of extranodal sites depends upon the organ involved and whether there is a known diagnosis of cHL. Identification of typical Hodgkin and Reed-Sternberg cells in needle biopsies of liver and marrow biopsies are not required because the specimens for evaluation are typically very small.
THERAPY
Related posts:
 Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


