Hodgkin lymphoma
Carol S. Portlock, MD  Anita Kumar, MD
Anita Kumar, MD  James Armitage, MD
James Armitage, MD
Overview
This chapter provides a historical perspective on the marked advances in radiotherapy and chemotherapy that have contributed to significant improvements in the curability of Hodgkin lymphoma (HL) over the past several decades. The classic features and new insights into the epidemiology, biology, and pathologic characteristics of HL are described. With emphasis on the modern clinical approach to the staging, imaging, and treatment of HL, the chapter highlights how current management approaches, such as refined risk stratification, use of PET imaging, and the reduction of radiation field and dose, seek to limit long-term toxicities of therapy while maintaining excellent cure rates. Finally, the authors describe how the development of novel, biologically targeted therapies has contributed to significant changes in the therapeutic landscape for patients with relapsed, refractory HL.
Introduction
Classical Hodgkin lymphoma, cHL, is now potentially curable in at least 80% of patients. Well-defined diagnostic criteria, prognostic factors, improved imaging, emphasis on systemic therapy first, and selective limited field radiotherapy consolidation have all contributed to this outcome. An increased appreciation of curability/quality of survivorship has now added the important goal of reducing the intensity of initial treatment when possible, without compromising cure rates; and, such changes in chemotherapy and radiation dose/field have resulted in reduction of long-term toxicities such as infertility, cardiopulmonary toxicity, and secondary malignancy.
History
In his historic paper of 1832, “On Some Morbid Appearances of the Exorbant Glands and Spleen,” Thomas Hodgkin described the clinical history and postmortem findings of massive lymph node and spleen enlargement in patients studied at Guy’s Hospital, London.1 Hodgkin recognized that these patients had suffered from a disease that started in the lymph nodes located along the major vessels in the neck, chest, or abdomen. Sir Samuel Wilks, and later W.S. Greenfield described the microscopic lymph node appearance. Carl Sternberg, in 1898 and Dorothy Reed, in 1902, are credited with the first definitive microscopic descriptions of Hodgkin lymphoma.2, 3 The early treatment of Hodgkin lymphoma with crude X-rays in 1901 followed the discovery of radiographs by Roentgen, radioactivity by Becquerel, and radium by the Curies at the end of the nineteenth century. Before this time, serum and other biologic preparations, arsenic, iodine, and surgery were ineffective in cHL. These first reports of successful X-ray radiograph treatments produced great excitement and premature predictions for the curability of Hodgkin lymphoma.4, 5
Modern radiation therapy (RT) techniques began in the 1920s with the work of Gilbert, who was one of the first to point out the predictable clinical patterns of cHL. He advocated treatment of apparently uninvolved adjacent lymph node chains that might contain suspected microscopic disease in addition to the involved nodal sites.6 In 1950, Vera Peters extended this approach, reporting 5- and 10-year survivals of 88% and 79%, respectively, for patients with stage I. 7 However, the concept that early-stage Hodgkin lymphoma might be curable with RT was slow to be accepted. Henry Kaplan developed the linear accelerator and successfully applied this technology to curing cHL. He defined radiation field sizes and doses, refined and improved diagnostic staging techniques, developed models for translating laboratory findings into clinical practice, and, with Saul Rosenberg, promoted early randomized clinical trials in the United States.8–10
Advanced-stage cHL was uniformly fatal until the development of combination chemotherapy. Mechlorethamine was shown to be an active drug in the 1940s, and in the mid-1960s Vincent DeVita and colleagues first treated patients with an effective four-drug regimen termed MOPP (mechlorethamine, vincristine, procarbazine, and prednisone).11 MOPP provided a high rate of complete remission and prolonged survival, resulting in curative outcomes.12
Like radiotherapy, randomized trials were also pivotal to the development of combination chemotherapy. These studies demonstrated no benefit of maintenance chemotherapy, established that treatment durations could be relatively short, and, ultimately, showed that ABVD (adriamycin, bleomycin, vinblastine, and dacarbazine) was associated with reduced toxicity and similar efficacy compared to MOPP (or MOPP-containing regimens), thus confirming ABVD as the standard regimen.13, 14
Epidemiology and etiology
In 2014, there were expected to be 9190 new cases of cHL in the United States. The incidence is 2.7 per 100,000 per year and has remained relatively constant for decades. In the United States, the median age for all new cases is 39 years with 31% occurring between the ages of 20 and 34. Somewhat more men than women (3.1 to 2.4 per 100,000) develop HL.15 Although a bimodal distribution was previously apparent in economically developed countries, the second peak is less apparent but persistent, as better pathology classification has found many of these cases to be non-Hodgkin lymphomas.
The etiology of Hodgkin lymphoma remains unknown. Epidemiologic data suggests both infectious and genetic components.16, 17 A viral etiology is suggested by an association between cHL in younger patients and childhood factors that decrease exposure to infectious agents at an early age, including increased maternal education, decreased numbers of siblings and playmates, early birth order, and single-family dwellings in childhood for economically developed countries.18, 19 This association has led to the proposal that cHL appears to mimic a viral illness that has an age-related host response to infection (such as seen with polio and infectious mononucleosis). Supporting this theory is the infrequent occurrence of cHL in children younger than 10 years in economically developed countries.
Epstein–Barr virus (EBV) is the leading viral candidate.16, 20, 21 EBV is the causative agent in African Burkitt lymphoma, and EBV-associated lymphomas are documented in patients with immune deficiency disorders and following organ transplantation. There is a two- to threefold excess in the incidence of cHL among patients with a prior history of mononucleosis. In addition, there is an altered antibody response pattern to EBV in patients who later develop cHL.
Recent cellular and molecular biology data have provided additional support for the association of EBV and cHL. Through the use of sensitive molecular probes, 30–50% of Hodgkin lymphoma specimens have been found to contain EBV genome fragments in the diagnostic Reed–Sternberg (R–S) cells.22 EBV genome status appears to be stable over time when studied in initial biopsies and at relapse. EBV genome-positive R–S cells express the so-called type II latency profile, with expression of latent membrane protein (LMP)-1, LMP-2a, EBNA-1, and EBV-encoded ribonucleic acid (EBER). LMP-1 is critical in transformation and acts as an oncogene in transfection studies, whereas EBNA-1 is essential for the replication of the episomal viral genome.
A genetic predisposition is evident with increased incidence among first-degree relatives, in some sibling studies, in monozygotic twins, and among parent–child pairs but not among spouses. In addition, cHL has been linked with certain human leukocyte antigens (HLAs) that are associated with EBV status; and genome-wide association studies have identified, but not proven, non-HLA susceptibility genes.17
Immunologic abnormalities in patients
cHL is characterized by functional deficits in cellular immunity and in T-cell-mediated immune responses that exist before treatment. These deficits persist in cured patients23 and include impairment of delayed cutaneous hypersensitivity, depressed proliferative responses to T-cell mitogen stimulation, enhanced immunoglobulin production, and decreased natural killer cell cytotoxicity. These abnormalities suggest an immunosuppression secondary to chronic overstimulation by cytokines. In patients with active cHL, these findings are consistent with increased cytokine secretion by R–S cells. However, it has been difficult to explain the persistence of these abnormalities in patients after successful treatment.
Treatment-induced immunosuppression returns toward normal after treatment, but has its greatest effect over the first few years.23 For example, there is an excess of herpes zoster infections, and more than 75% of such cases occur within the first year. Few occur after the third year (6%). The risk of H. zoster appears highest as the intensity of therapy increases.24
Unlike the deficits in delayed hypersensitivity, most patients with cHL at diagnosis appear to have relatively normal B-cell number and function which may be adversely affected by treatment.23 Patients should be encouraged to keep vaccinations up to date.
Pathology
Hodgkin lymphoma is unique among lymphomas for its histologic diversity.25 Involved lymph nodes contain varying degrees of normal reactive and inflammatory cells, fibrosis, and a scattering of the characteristic malignant cHL cells, the R–S cells, and their mononuclear variants. The typical R–S cell has abundant cytoplasm and 2–3 nuclei, each with a single prominent nucleolus. The large size and unusual appearance of the R–S cell sets it apart from the adjacent smaller background cells. The mononuclear variants have nuclear and cytoplasmic features of R–S cells, but have only a single nucleus. Although the diagnosis of cHL should rarely be made in the absence of R–S cells, the presence of these cells alone is not sufficient to make the diagnosis. R–S-like cells have been found in infectious mononucleosis, non-Hodgkin lymphoma, and in some carcinomas and sarcomas. Thus, criteria for a cHL diagnosis include the presence of the R–S cells and the characteristic background of normal lymphocytes, plasma cells, and eosinophils.
The current pathology classification includes cHL: nodular sclerosis (NSHL), mixed cellularity (MCHL), lymphocyte-rich (LRHL), and lymphocyte depletion (LDHL) subtypes; as well as NLPHL, nodular lymphocyte predominance Hodgkin lymphoma. The subtyping of cHL does not affect clinical management, prognosis, or therapy. Nevertheless, the histologic subtypes are associated with different presentations, distinct natural histories, and variable prognoses. These differences are most evident in NLPHL.
Hodgkin lymphoma subtypes
Two histologic features of NSHL help to differentiate this cHL subtype: a proliferation of collagenous bands dividing the lymph node into circumscribed nodules and these nodules contain a variant R–S cell called the lacunar cell. In formalin-fixed tissue, this cell’s abundant pale cytoplasm often retracts and gives the appearance of a cell in space (Figures 1–3). Molecular profiling studies have shown a close relationship between primary mediastinal large B-cell lymphoma and NSHL, rarely a “gray-zone” neoplasm that combines features of both neoplasms may be identified.26–28
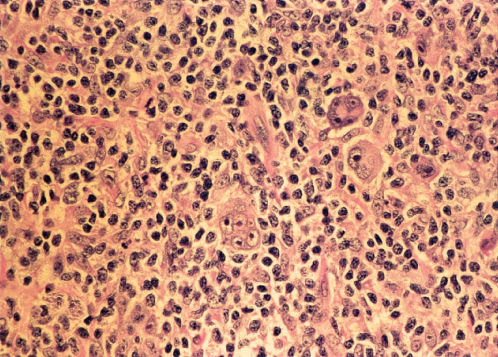
Figure 1 Reed–Sternberg cells and variants in Hodgkin lymphoma of the nodular sclerosis type. Large multinucleated or multilobated cells and a few mononuclear cells with macronucleoli stand apart from cellular background elements.
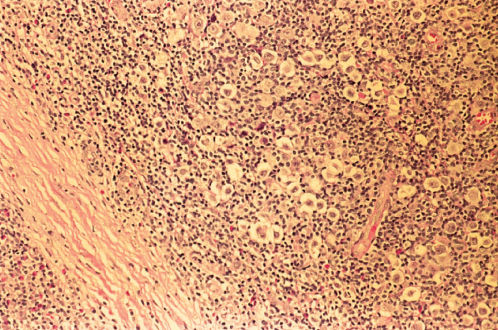
Figure 2 Hodgkin disease, nodular sclerosis. A fibrous band is present in the left lower part of the field. Neoplastic lacunar cells having abundant, clear cytoplasm stand out against the lymphocytic background.
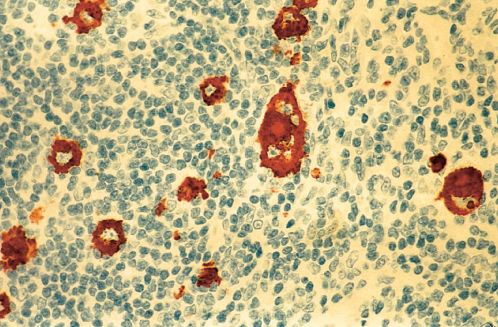
Figure 3 Immunostain for CD15 in Hodgkin disease, nodular sclerosis type. Neoplastic Reed–Sternberg cells and mononuclear Hodgkin cells show positive immunoreactivity.
NSHL is the only subtype of cHL as common in women as in men. It occurs in adolescents and young adults and is unusual in patients older than 50 years. It has a striking propensity to involve lower cervical, supraclavicular, and mediastinal lymph nodes with an orderly pattern of spread.29 It makes up 60–70% of cHL in economically developed countries, but is less commonly seen in underdeveloped countries.
MCHL has an inflammatory background abundant in normal cells as well as 5–15 R–S cells and variants per high-power field (Figures 4 and 5). These patients are older, more likely to have B symptoms, and often have abdominal involvement or advanced disease. Approximately 25% with cHL in the United States have MCHL, and it is more common in underdeveloped countries. MCHL can be confused with peripheral T-cell lymphoma, and the antigen PAX5 may be particularly helpful, as it is a B-cell marker.
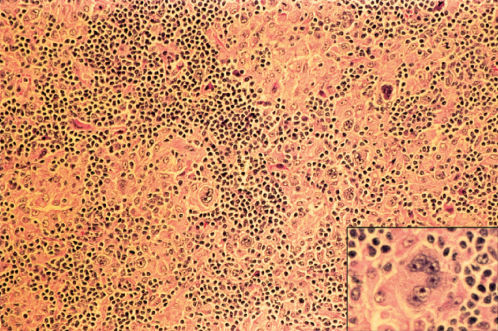
Figure 4 Hodgkin disease, mixed cellularity type. Reed–Sternberg cells in histiocyte-rich cellular background. Inset: Multinucleated Reed–Sternberg cell at higher magnification.
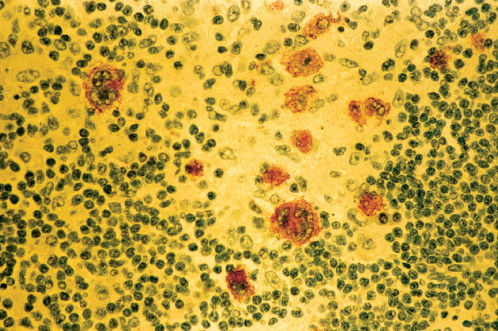
Figure 5 Immunostain for Epstein–Barr virus-latent membrane protein in Hodgkin disease, mixed cellularity type (same biopsy as Figure 4).
LRHL may resemble other histologic subtypes and may be nodular or diffuse. R–S cells are relatively rare, and the background is dominated by small mature lymphocytes (Figures 6 and 7). Eosinophils and neutrophils are usually restricted to blood vessels.
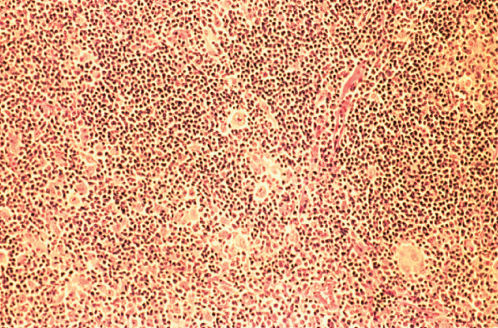
Figure 6 Lymphocyte-rich classic Hodgkin disease. Reed–Sternberg cells and mononuclear Hodgkin cells are relatively rare within the background proliferation of small lymphocytes and histiocytes.
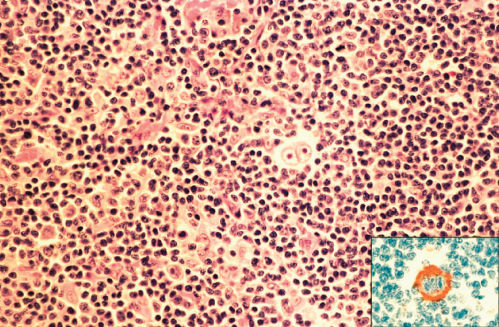
Figure 7 Lymphocyte-rich classic Hodgkin disease. Binucleated Reed–Sternberg cell in center of field. In same biopsy, Reed–Sternberg cells immunostained positively for CD15 (inset).
LDHL is rarely diagnosed, accounting for less than 1% of cHL in economically advanced countries. Generally, these patients present with advanced disease and B symptoms. R–S cells and “pleomorphic” variant cells are frequent and most cases have only sparse normal lymphocytes.
In NLPHL, the lymph node architecture is usually effaced, although a remnant of normal nodal architecture may remain. Diagnostic R–S cells are not seen, but variant lymphocyte predominance (LP) cells are typical (Figures 8 and 9). These cells often have multilobated nuclei and have been called popcorn cells because of their resemblance to a popped kernel of corn. Fibrosis is not usually seen. The LP or “popcorn” R–S variants occur in a background of polyclonal B lymphocytes (Figure 9).30, 31 LP cells are usually positive for the B-cell marker CD20, but negative for the cHL markers of CD15 and CD 30 (Figures 10 and 11).31, 32 EBV is rarely detected in NLPHL.33 The benign disorder, progressive transformation of germinal centers, is often associated with NLPHL. R–S cells and LP variants are absent in this entity. Progressive transformation of germinal centers can also be seen in association with NLPHL in the same or an adjacent node.32
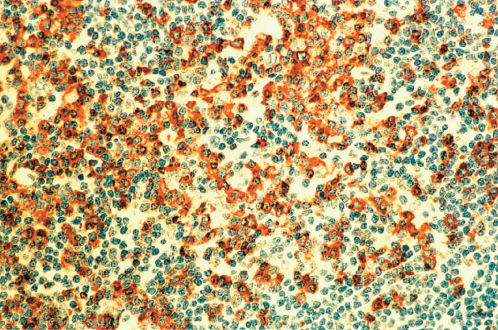
Figure 8 Lymphocyte predominant Hodgkin lymphoma (same biopsy as Figures 9–11). Immunostain for CD57 reveals a marked increase in immunoreactive cells showing localization around nonimmunoreactive L and H cells within a nodule. The CD3 immunostain showed a similar distribution of immunoreactive cells.
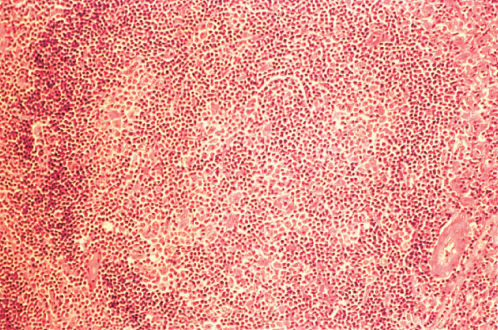
Figure 9 Lymphocyte predominant Hodgkin disease. The vaguely nodular histologic pattern is apparent.
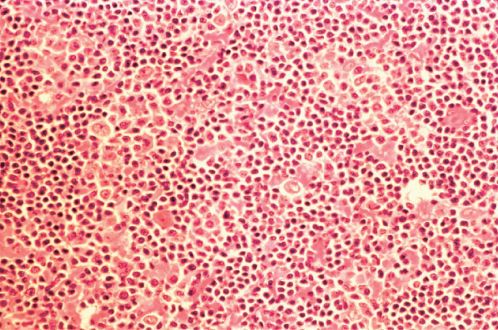
Figure 10 Lymphocyte predominant Hodgkin lymphoma at higher magnification (same biopsy as Figure 9). Within the background of lymphocytes and histiocytes are scattered large lobated cells having a fine chromatin pattern, relatively small nucleoli, and sparse cytoplasm so-called L and H cells.
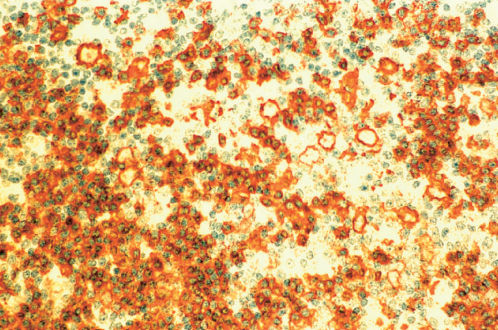
Figure 11 Lymphocyte predominant Hodgkin lymphoma (same biopsy as Figures 9 and 10). Immunostain for CD20 demonstrates positive staining of L and H cells as well as a high percentage of lymphocytes within a nodule.
NLPHL makes up 5–10% of all Hodgkin lymphoma in the United States. It is often localized to a single peripheral nodal region (high cervical, submandibular, epitrochlear, inguinal, or femoral nodes) and infrequently involves mediastinal or abdominal sites.
Immunophenotype and biology
The R–S cell is of B-lymphocyte origin.25 These cells may express antigens found on resting or activated lymphocytes, most often B cell: usually positive for PAX5, and infrequently, CD20 or CD79a; and rarely positive for T-cell surface antigens (CD3, CD4, and CD8). The LP cells of NLPHL consistently express B-cell antigens. In cHL, the surface antigens CD30 and CD15 are present on most R–S cells, and nearly all cases are CD30+, and approximately 80% are CD15+, whereas CD45 (leukocyte common antigen) is usually negative. In contrast, the LP cells of NLPHL are CD15 and CD30 negative and CD45 positive.33
Microdissection studies of single R–S cells demonstrate clonal immunoglobulin gene rearrangements in the vast majority of cases, confirming the clonality of the cells and establishing their B-cell lineage, despite their aberrant lack of immunoglobulin gene expression.34, 35 Moreover, R–S cells possess somatically mutated V genes, implying a germinal center or postgerminal center origin. The mutational pattern suggests that this may account for the lack of immunoglobulin expression. Other possible mechanisms include epigenetic silencing of heavy chain gene transcription or constitutive expression of Notch 1 and STAT 5.25 Normal germinal center B cells that lack functional immunoglobulin receptors are usually eliminated within the germinal center via apoptosis; and therefore, R–S cells appear resistant to this usual apoptotic mechanism. This apoptosis pathway via FAS/CD95 is inhibited by c-FLIP, a gene that is constitutively expressed by R–S cells.36 Several hypotheses have been generated to explain this phenomenon, including activation of the NF-κB pathway, general lineage promiscuity, and Epstein–Barr infection. Evidence favoring the NF-κB pathway includes the observation of constitutive expression of NF-κB in Hodgkin lymphoma-derived cell lines, the finding that suppression of NF-κB impairs tumor growth in severe combined immunodeficient mice and growth of cell lines,37 and an epidemiology study showing that regular aspirin use is associated with a reduced risk of developing Hodgkin lymphoma, presumably through inhibition of NF-κB transcription.38 The cause of the constitutive activation of NF-κB may include amplification of the REL gene, mutations in NF-κB inhibition, and somatic mutations in TNFA1P3.25 LMP-2a expression may substitute for a functional B-cell receptor in EBV-associated cases.
Microdissection studies of LP cells also demonstrate clonal immunoglobulin gene rearrangements, again establishing clonality and confirming B-cell lineage.34 In addition to demonstrating somatically mutated V genes, intraclonal sequence diversity can also be detected, providing strong evidence that NLPHL is a germinal center lymphoma. In contrast to cHL, the mutations are compatible with functional antigen receptors.
Cytogenetic abnormalities are common in R–S cells; however, no consistent pattern has been described. Rarely, the t(14 : 18) translocation, common in follicular B-cell lymphomas, may be detected. Comparative genomic hybridization studies demonstrate recurrent gains on chromosomal arms 2p (the site of NF-κB, REL, and BCL1 1a), 9p (the site of JAK2), and 12q (the site of MDM2), and amplifications on chromosomal bands 4p16, 4q23–q24, and 9p23–p24 (associated with high level of PD-L1 protein expression). Recurrent imbalances have been demonstrated in a majority of cHL using genome-wide GeneScan technology.39
The tumor microenvironment has been found to have an important role in the pathogenesis, associated manifestations, and prognosis of Hodgkin lymphoma.40, 41 It has been hypothesized that cytokines are responsible for the marked inflammatory component, fibrosis, and diverse histologic patterns of cHL, as well as the associated clinical symptoms of fever, weight loss, and night sweats.42–44 Many cases are associated with upregulation of tumor necrosis factor receptor (TNFR) and ligand family members, Th2 and to a lesser extent Th1 cytokines, and other chemokines. Necrosis factor receptor (NFR) members may lead to constitutive activation of NF-κB, an important factor in proliferation and survival of B lymphocytes. Preferential expression of Th2 cytokines and chemokines may explain the frequent presence of eosinophils and fibroblasts, as well as local suppression of the cellular immune response. EBV may contribute to the production of cytokines, for example, via LMP-1-induced activation of NF-κB and stimulation of interleukin (IL)-10, a potent inhibitor of cellular immunity. In some cases, specific cytokines may be associated with specific histologic features. For example, transforming growth factor (TGF), a known stimulus for fibroblast proliferation and collagen formation, is associated with NSHL,45 and TARC (CCL27), a lymphocyte-directed CC chemokine secreted by Hodgkin cells, may be responsible for the infiltration by CD4+ T cells.46 Tissue eosinophilia may be due to expression of IL-5, IL-9, CCL11, and CCL28; and IL-13 may play a role in autocrine stimulation of R–S cells.47
Staging
Most patients with cHL have a central pattern of lymph node involvement (cervical, mediastinal, and paraaortic) with >80% presenting initially above the diaphragm. In contrast, certain nodal chains (mesenteric, hypogastric, presacral, epitrochlear, and popliteal) are seldom, if ever involved. Spleen involvement is associated with adenopathy below the diaphragm and systemic symptoms. Isolated liver disease is rare; bone and bone marrow involvement is usually focal. Staging has recently been updated with the Lugano classification, incorporating the original Ann Arbor Stage and the later Cotswolds revision (Table 1).48 PET imaging is now recognized as the most accurate means of identifying all sites of cHL involvement, and routine bone marrow biopsy is no longer recommended. See Table 2 for pretreatment evaluation.
Table 1 The Lugano classification.48
| Stage I | A single lymph node or a group of adjacent nodes. Stage IE: A single extralymphatic site Tonsils, Waldeyer’s ring, and spleen are considered nodal tissue |
| Stage II |