Chapter 28 HIV Resistance Testing in Clinical Practice
INTRODUCTION
During the last decade our understanding of both the biology and the clinical relevance of human immunodeficiency virus (HIV) drug resistance has increased exponentially. There is now evidence that emergence of drug resistance to the initial highly active antiretroviral therapy (HAART) regimen is associated with an increased risk of death.1 If drug resistance emerges to all three major drug classes (nucleoside reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors (NNRTIs) and protease inhibitors (PIs)), the risk of death is substantially increased.2
BIOLOGY OF DRUG RESISTANCE
Evolution of Drug Resistant Viruses
In the past it was generally believed that drug resistance was an unavoidable consequence of antiretroviral (ARV) therapy.3 This assumption was based on the knowledge that HIV replication is error prone (roughly one transcription error per every 4 Kb of transcribed cDNA), the lack of transcription proofreading enzymes, and the production of billions of viral particles every day (see Chapter 4). Thus, all infected individuals have potential to quickly develop an extremely heterogeneous viral population with a seemingly unlimited number of variants. As a consequence, all possible resistance patterns theoretically exist as subpopulations in untreated patients. Exposure to ARV pressure, therefore, has potential to select for the preexisting resistant subpopulation. This concept was derived from modeling based on HIV variability in initial treatment studies using suboptimal regimens such as single or double nucleoside combinations.
Mutations readily occur in HIV when patients are treated with HAART regimens that are not optimally suppressive. However, the resulting drug-resistant viruses do not necessarily have equal replicative capacity (RC) in vitro when compared to wild-type virus. Thus, resistant virus may not overtake wild-type virus, allowing some resistant viruses to remain as minority variants. This suggests that, in such settings, retaining drug therapy even when viral load value is above 50 copies/μL may be beneficial since the resistant virus may never achieve the viral loads that would be encountered if the wild-type repopulates as the dominant species. The less fit virus populations result in viral load values that are significantly below the pretherapy baseline level created by wild-type virus. An example of this phenomenon is the emergence of the M184V-resistant mutant selected by lamivudine monotherapy. Once this mutation appears in the viral population while patients are on lamivudine monotherapy, the HIV RNA level rebounds toward baseline but remains 0.6–0.8 log below pretherapy levels. This phenomenon of partial suppression is best explained by the classical Darwinian principle of survival of the fittest. Drug-resistant variants, preexisting as minority species owing to a reduced replicative capacity compared to wild-type virus, become dominant under conditions of drug selection pressure while the wild-type virus is suppressed by the antiviral regimen. Thus, the less fit virus predominates during therapy, but might not attain the same level of replicative success as the wild-type virus.
Resistance Mechanisms
Other more complex mechanisms also exist. Several common resistance-conferring mutations to NRTI agents can improve the enzymatic efficiency of the reverse transcriptase (RT)4,5 enzyme, thereby enabling the RT to more efficiently remove the terminal nucleoside triphosphate incorporated into a growing DNA strand (excision).6,7 In other words, the RT enzyme with the resistance-conferring mutations more efficiently “throws out” any unnatural nucleoside, i.e., the drug. This mechanism results in resistance to a broad number of nucleoside agents. An example of this type of resistance mechanism is the development of the K65R mutation.
Novel mechanisms leading to resistance to PI drugs have been identified. Changes in the PI substrate, e.g., the cleavage site, can lead to resistance to the PIs in the absence of changes in the protease itself.8 The clinical importance of this mutation is under investigation.
Mutational Patterns and Hypersusceptibility
For most drugs, the development of resistance occurs through accumulation of mutations that lead to progressively reduced drug susceptibility. The mutations have an additive or synergistic resistance effect.9 Generally the chance of cross-resistance to other drugs in the same class increases as the number of mutations grows.
“Thymidine-associated mutations”, or TAMs pathways, are the best described pattern of mutation accumulation relevant to the thymidine analog nucleoside drugs. The most common TAMs are M41L, D67N, K70R, T215Y/F, L210W, and K219Q/E. These TAMs confer resistance not only to the drugs that select for them (zidovudine and stavudine) but also confer cross resistance to most other NRTIs, such as didanosine, tenofovir, and abacavir. Cross resistance to TAMs is critically dependent on the number of mutations. The more TAMs, the more resistance there is to more nucleoside drugs.
GENOTYPIC ASSAYS
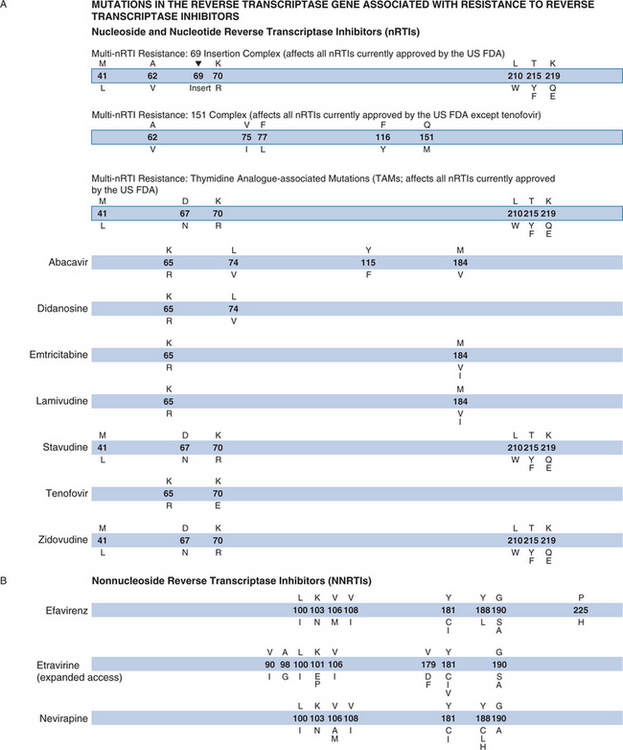
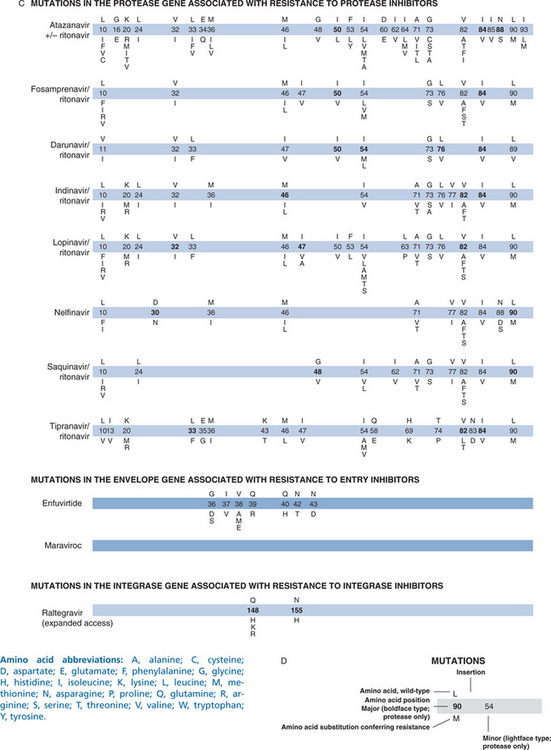
Given the complexity of these assays, the sensitivity and specificity of assays performed by individual laboratories must be carefully and regularly assessed.10,11 Genotypic assays are not easy to interpret when multiple mutations are present simultaneously. Summary tables and on-line resources are useful for interpreting complex patterns. Most laboratories provide interpretation in their patient result reports (Fig. 28-1).9 Despite this complexity, many clinicians are learning how to incorporate genotypic information into their treatment strategies.
PHENOTYPIC ASSAYS
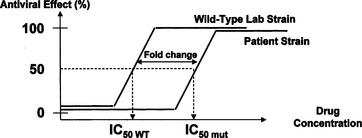
Most commercial phenotypic assays are recombinant virus assays that amplify the RT, protease, integrase and segments of the envelope genes from the predominant quasispecies of the viral RNA in the patient’s plasma. These amplified fragments are inserted into a laboratory virus strain which lacks these genes. The resulting virus is then permitted to replicate in cell culture in the presence of various drug concentrations (Fig. 28-2). Viral replication is detected by emission of light induced by the activity of a luciferase gene product introduced into the laboratory viral strain as an indicator. Results are expressed as the concentration of drug required to inhibit 50% of growth (IC50) and as the fold change in IC50 relative to a wild-type control strain.




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


