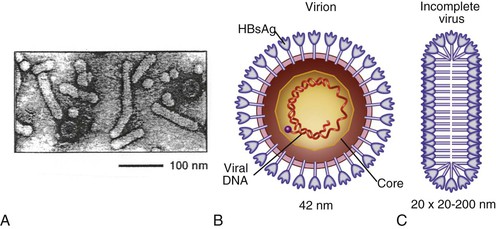
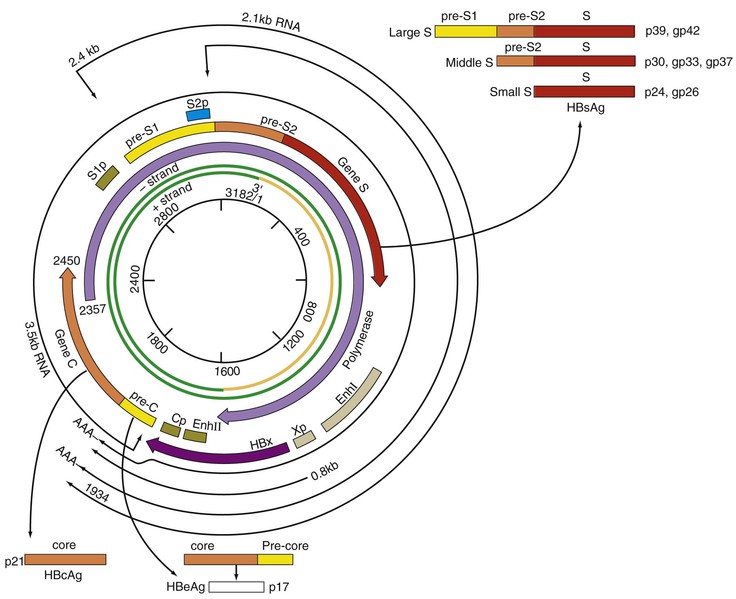
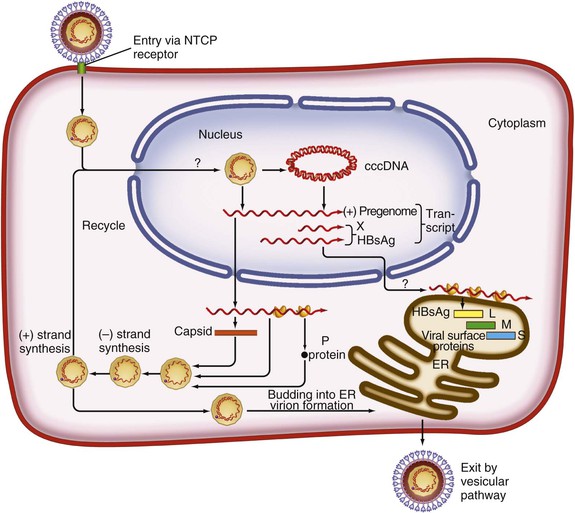
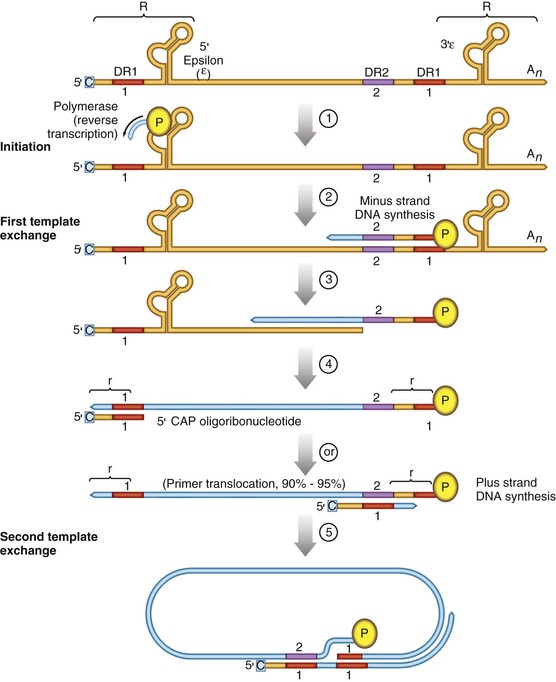
Chloe Lynne Thio, Claudia Hawkins Hepatitis B virus (HBV) infects more than 500 million people worldwide. It is the leading cause of chronic hepatitis, cirrhosis, and hepatocellular carcinoma (HCC), and these sequelae of chronic infection account for more than 1 million deaths annually. The outcome of infection and spectrum of illness varies widely. During the acute phase, infections range from asymptomatic hepatitis to icteric hepatitis, including fulminant hepatitis. Once chronic infection is established, the spectrum of illness ranges from the asymptomatic, healthy carrier state to progressive liver disease that results in the sequelae of end-stage liver disease, including cirrhosis and HCC. Even though HBV cannot be cultured easily, enough is known about the viral life cycle that specific antivirals are available to control viral replication in patients; however, no cure is available yet for hepatitis B. The identification of the correlates of protective immunity has facilitated the widespread use of a highly effective vaccine, which by preventing chronic carriage of the virus and the subsequent development of HCC represents the first true cancer vaccine. Hippocrates recognized the spread of jaundice by infectious agents as early as 4000 bc. Recently, the oldest known HBV was isolated from the liver of a mummified Korean child from the 16th century, which is estimated to have originated between 3000 and 100,000 years ago.1 The early modern cases of HBV infection were linked to the use of conventional viral vaccines, which were prepared from or contained human serum. In 1885, Lurman described the appearance of jaundice in 15% of 1289 shipyard workers who received smallpox vaccine prepared from human lymph.2 Epidemics of hepatitis were also recorded following the administration of yellow fever vaccine, which was stabilized with human serum.3 In the early part of the 20th century, the increasing use of contaminated syringes and needles by diabetics on insulin and by patients treated for syphilis at venereal disease clinics elevated the importance of serum hepatitis.4,5 This led to the association of hepatitis B with blood and blood products, as well as to its distinction from infectious hepatitis, caused by hepatitis A virus, a member of the Picornaviridae family.6 The first hint of viral etiology came from the studies of Blumberg, who reported the discovery of a human antigen in Australian Aboriginals termed Australian (Au) antigen.7 Subsequently, the Au antigen came to be known as hepatitis B surface antigen (HBsAg), and its association with acute hepatitis was established. For this discovery, Dr. Baruch Blumberg received the Nobel Prize in Physiology and Medicine in 1976. In 1971, Dane, an electron microscopist, visualized the presence of 22-nm HBsAg subviral particles along with the complete 42-nm virus particles in the blood of hepatitis B patients (Fig. 148-1).8 Due to their unique biologic and molecular characteristics, as well as their liver tropism, HBV and HBV-like animal viruses were given the status of a new family designated Hepadnaviridae (hepatotropic DNA viruses).9 HBV, the human pathogen, and other mammalian hepatitis viruses with sequence homology and similar genome organization are grouped in the genus Orthohepadnavirus. The genus Avihepadnavirus includes viruses that infect ducks, geese, and heron. The Hepadnavirus animal models, which include duck hepatitis B virus,10 woodchuck hepatitis virus, and ground squirrel hepatitis virus, have been extensively studied and have contributed to the current knowledge of the molecular biology of HBV infection and replication. However, these viral genomes are considerably divergent from human HBV, and there are limited tools to study the immune reaction to the virus in these animals. The primary host for HBV is human, but HBsAg has been detected in other primates such as chimpanzees, gibbons, orangutans, African green monkeys, and squirrel monkeys. However, the use of these animals for the study of pathogenesis is both difficult and expensive. Thus, a convenient animal model or an efficient tissue culture system for HBV remains unavailable. HBV was molecularly cloned from patients’ sera in 1979 and its complete DNA sequence determined.11–13 These advances led to a surge of investigations on the molecular aspects of HBV molecular biology, chief among which are the regulatory schemes of gene expression and replication. HBV is a small DNA virus, whose 3200 kilobase (kb) partially double-stranded DNA genome is maintained in a circular conformation.14 Partially duplex DNA molecules represent incomplete synthesis of the viral DNA during morphogenesis (Fig. 148-2).15 One of the unique features of HBV infection is the production of large quantities of subviral spherical and filamentous HBsAg particles in addition to complete virus particles. Under electron microscopy, these are distinguished by a diameter of 42 nm for complete virus (Dane) particles and 22-nm spherical and filamentous structures for subviral particles (see Fig. 148-1).8 HBsAg is expressed on the exterior of all these particles. HBsAg subviral particles reach a titer of 1013/mL, whereas viral particles range in titer from 104/mL to 109/mL. All of these particles circulate in blood and permit convenient diagnosis of viral antigen by enzyme-linked immunosorbent assay (ELISA) or radioimmunoassay (RIA).16 HBsAg forms the viral component of the lipoprotein envelope, which encloses a core shell containing the viral DNA genome and the virus-encoded polymerase protein. The nucleocapsid or core is composed of a 21 kDa basic phosphoprotein commonly known as hepatitis B core antigen (HBcAg).17 A cell-derived kinase activity has been shown to be associated with the virion particles,18 but the functional significance of this enzyme is not understood. The 22-nm HBsAg particles and filaments are composed of three forms of surface antigen polypeptides and lipids derived from hepatocyte membranes. The lipid content is approximately 30% by weight and includes phospholipids, cholesterol, cholesterol esters, and triglycerides.19,20 These particles are devoid of HBV DNA genome and hence are noninfectious. Because of their high immunogenicity, purified HBsAg particles can be used as an HBV vaccine. HBsAg elicits neutralizing antibodies, which offer protection from reinfection.21,22 The high titers of HBsAg in patients during natural infection can potentially serve to adsorb neutralizing antibody and thus protect the virus from host defenses. HBV primarily infects hepatocytes. The pre-S1 domain of the large HBsAg protein promotes attachment and entry of HBV into the hepatocyte via liver cell–specific receptor recently identified as Na (sodium) taurocholate cotransporting polypeptide (NTCP), which is an integral membrane protein used in bile acid transport.23,24 There is evidence that hepatocyte entry may be a multistep process including binding to heparan sulfate proteoglycans,25 which are found on a variety of cells, and clathrin-mediated endocytosis.26 Bound virions deliver core particles into the cytoplasm, which make their way into the nucleus through the nuclear pore complex.27 In the nucleus, the virion DNA, which is partially duplex, is matured into a covalently closed circular (ccc) DNA form (Fig. 148-3).10 Liver specificity is also displayed at the level of viral gene expression, which is controlled largely by the promoters and enhancers (see Fig. 148-2).29 HBV uses unique transcriptional and translational strategies to maximize the limited coding capacity of its genome. The HBV DNA genome is a partially double-stranded molecule within the virions (see Fig. 148-1).30 The complete (negative) strand contains a protein covalently linked to its 5′ terminus.31 The incomplete (plus) strand displays variable length and bears a 5′ capped oligoribonucleotide at its 5′ end (see Fig. 148-2).32 The asymmetry of the DNA strands reflects the incomplete synthesis of DNA during maturation of viral particles. HBV DNA codes for four overlapping open reading frames (ORFs): S, for the surface antigen or envelope gene (HBsAg); C, for the nucleocapsid (core) and “e” antigen gene (HBcAg, HBeAg); P, for the polymerase gene; and X, for the HBx gene (see Fig. 148-2). The surface antigen ORF contains three in-frame initiator codons from which small or major (S), middle (M), and large kDa (L) HBsAg polypeptides are synthesized, respectively. These polypeptides are variably glycosylated to yield several species: S, p24/gp26; M, p30/gp33/gp37; and L, p39/gp42 kDa HBsAg polypeptides, respectively. L and M HBsAg contain characteristic pre-S1 and pre-S2 domains, and hence these proteins are also referred to as pre-S1 and pre-S2 proteins, respectively. The 22-nm subviral particles are composed of mostly S and lesser amounts of M polypeptides and few or no L polypeptides. The 42-nm Dane particles, which represent the complete virus, contain all three HBsAg forms. The C region or the core ORF contains two in-frame initiator codons with the first ATG start codon responsible for e antigen polypeptide synthesis (HBeAg) (see Fig. 148-2). The second ATG serves as an initiation codon for the HBcAg polypeptide. HBeAg is secreted from cells and accumulates in serum as an immunologically distinct soluble antigen and serves as a marker of ongoing viral replication.33 Both gene products (core and e polypeptides) are made from the same reading frame. The P and X ORFs encode the polymerase and the HBx proteins, respectively. Before the advent of current molecular methods, HBV was divided into serotypes on the basis of the antibody response to HBsAg. However, HBV is now divided into 10 genotypes (A-J) based on genetic diversity of at least 8% in the HBV genome.34,35 Within these genotypes are subgenotypes, which differ by a minimum of 4%. These genotypes and subgenotypes are distributed geographically with genotypes A in North America, Europe, and parts of Africa; genotypes B and C in Asia; genotype D in India, the Middle East, the Mediterranean region, and parts of Africa; genotype E in Africa; genotype F in Central and South America; genotype G in France, Germany, and North America. Genotype H is found in Central America. Genotype I, a novel recombination between genotypes A, C, and G, is found in Vietnam and Laos.36 Genotype J was identified in Japan.37 There are accumulating data that these genotypic differences are associated with disease and treatment outcomes. Genotypes G and C are associated with more severe liver disease and higher HCC risk.38 Genotype C also has slower HBeAg seroconversion compared with genotype B.39–41 Genotypes A and B are more responsive to pegylated interferon-α (PEG IFN-α) therapy than are genotypes C and D.42 The HBV genome maximizes the use of its limited genomic size by using multiple overlapping reading frames and multiple initiation codons to generate antigenically different proteins. The covalently closed circular DNA (cccDNA) in the nucleus serves as the substrate for viral transcription and uses host polymerase II (pol II) in the synthesis of viral transcripts (see Fig. 148-3). The four different genes, designated C, S, P, and X, encode HBc/eAg, HBsAg, polymerase, and HBx proteins, respectively.16 The expression of these genes is regulated by four promoter elements (S1p, S2p, Cp, and Xp) and two enhancer elements (enhancer I and II; see Fig. 148-2). These transcriptional regulatory elements direct the synthesis of multiple viral transcripts that are approximately 3.5, 2.4/2.1, and 0.8 kb in length, respectively. All viral transcripts are unspliced, capped, and polyadenylated. All the transcripts are encoded on one strand of the DNA and co-terminate at an identical polyadenylation site.43 The pre-S1 (S1p) promoter directs the synthesis of the 2.4-kb transcripts, which code for the large envelope (L HBsAg) polypeptide. This promoter contains binding sites for two liver-enriched transcription factors, HNF-3 and HNF-1, which are primarily responsible for the liver-specific activity of this promoter.44 The pre-S2 or S (S2p) promoter directs the transcription of multiple species of messenger RNAs (mRNAs) coding for pre-S2/M and S HBsAg polypeptides. Collectively these RNA species are approximately 2.1 kb in length with 5′ heterogeneous ends. The mRNAs, which initiate downstream of the pre-S2 initiator ATG, code for the S, or the major polypeptide. The S2p promoter also displays liver specificity and is controlled by the enhancer II element located about 2000 bp away (see Fig. 148-2).45 The S2p promoter is stronger than the S1p promoter, which results in the synthesis of an excess of the major S over the L (pre-S1) and M (pre-S2) forms of the HBsAg. This regulation is especially critical for synthesizing the appropriate levels of the three forms of surface proteins within the cell. The basis for the differential regulation of these promoters is governed by mechanisms that involve both positive and negative cis-acting elements and the trans-acting transcriptional factors.16,46 The core/pregenomic promoter (Cp) governs the expression of two longer-than-genome-length transcripts (3.5 kb) designated pre-core (pre-C) and core (C) RNAs. The slightly longer pre-core RNA directs the translation of HBeAg polypeptide. The shorter C RNA is used for the translation of core and polymerase proteins. The ATG for the polymerase (reverse transcriptase) is located several hundred nucleotides from the 5′ end (see Fig. 148-2). The C RNA, after its translation into core polypeptides, packages its own RNA, which then functions as a pregenome RNA (pregenomic RNA). The Cp promoter contains binding sites for several liver-enriched and ubiquitous transcription factors.46 Immediately juxtaposed to this lies enhancer II, which in concert with enhancer I, plays a regulatory role in the overall HBV gene expression. A complex scheme of transcriptional regulation appears to operate within these control elements.47 The X promoter (Xp) is located immediately downstream of enhancer I and regulates the synthesis of the low abundance 0.8-kb X transcripts that correlate with similar levels of HBx protein synthesis in infected cells.48,49 Both ubiquitous and liver-enriched factors binding to enhancer I regulate the biosynthesis of X transcripts. HBV also uses unique translational strategies to maximize the limited coding capacity of its genome. HBV encodes three major viral transcripts discussed earlier (see Fig. 148-2). The S region contains three in-frame translational start codons from which the synthesis of three distinct surface antigen polypeptides is regulated (L/pre-S1, M/pre-S2, and S). These proteins exhibit distinct amino-termini and differ with respect to the extent in which they are post-translationally modified by glycosylation.50 The S protein is referred to as the major surface antigen because it represents approximately 85% of the HBsAg that is produced by the virus. The pre-S1 (L) and pre-S2 (M) proteins are present at an abundance of approximately 15% and 1% to 2%, respectively. The pre-C and C mRNAs are translated into e and core polypeptides (HBc/eAg), respectively. The pre-C protein contains a signal peptide sequence encoding 19 amino acid residues, which targets this protein to the secretory pathway in the endoplasmic reticulum (ER).51 Further processing of the protein includes cleavage of the signal peptide, and several amino acid residues at the carboxy-terminus leading to the production of a 17-kDa HBeAg (see Fig. 148-2). HBeAg is secreted and found in the serum of patients and serves as a marker of active replication in chronic hepatitis. Although the function of HBeAg is not clearly understood, one study demonstrated that it downregulated Toll-like receptor 2 expression on hepatocytes and monocytes leading to a decrease in cytokine expression.52 These data suggest a role for HBeAg in modifying the innate immune response to HBV. HBeAg is dispensable for replication, as HBV carriers with pre-C mutant viruses arise spontaneously and are both infectious and pathogenic.53,54 The C RNA initiates downstream of the pre-C ATG and translates into HBcAg and less frequently into polymerase. Because polymerase ATG is located several hundred nucleotides downstream of the 5′ end (see Fig. 148-2), the ribosomes must access this initiator codon by a mechanism different from ribosome scanning. Because this process is inefficient, one polymerase protein is translated for every 200 to 300 molecules of core polypeptides. HBV polymerase protein consists of 832 aa and is composed of four domains: terminal protein (TP), spacer, reverse transcriptase (RT), and ribonuclease H (RNaseH).16 The TP domain represents the portion of the polymerase protein that is covalently bound to minus-strand DNA. The spacer domain or tether region connects the TP domain to the RT, which contains the catalytic domain for the polymerase. The RT domain is followed by an RNaseH domain, which functions to hydrolyze the pregenomic RNA template after reverse transcription within the nucleocapsid.55,56 Although HBV is a DNA-containing virus, the replication occurs via an RNA intermediate, a characteristic that places hepadnaviruses close to retroviruses. Much of our understanding of HBV replication strategy comes from the classic experiments of Summers and Mason,57 which demonstrated that HBV amplifies via reverse transcription of an RNA intermediate and that these events occur within the subviral core particles in the cytoplasm. The complex mechanism by which pregenomic RNA is converted to partially double-stranded virion DNA begins with the encapsidation of pregenomic RNA by the core polypeptide along with the polymerase (Fig. 148-4).58,59 The pregenomic RNA contains terminally redundant 200 nucleotide sequences, which include an epsilon (ε) stem-loop structure and a short sequence of 11 to 12 nucleotides termed DR1 (see Fig. 148-4). The HBV polymerase binds pregenomic RNA preferentially at the 5′ copy of the ε stem-loop structure on its own molecule. The interaction between the ε signal of the pregenomic RNA and the polymerase and the core leads to the formation of a ribonucleoprotein complex. The HBV capsid or core assembles into a replication-competent particle.16 The sequence of events and the mechanism of assembly remain to be characterized. The TP domain of the polymerase protein functions as a protein primer for the initiation of reverse transcription in a process called nucleotide priming or protein-primed reverse transcription.60 The positive strand is extended to variable lengths to yield mature HBV DNA. The cessation of the positive strand synthesis at various stages of its synthesis coincides with the maturation of the core particles and their entry into the ER leading to the packaging of partially duplex genomic DNA found in hepatitis B (HB) virions (see Fig. 148-3).61 This entry prevents access to cytoplasmic deoxynucleotide pools needed for completion of DNA synthesis. Matured core particles face two options at this stage: they either enter the ER, undergo virion packaging with HBsAg polypeptides, and bud from the membrane as 42-nm Dane particles, or they enter the nucleus and deliver the partially duplex DNA and repeat the cycle of replication (see Fig. 148-3). In the nucleus, virion DNA is repaired to produce cccDNA molecules. Recent reports demonstrate that less than 1 copy of cccDNA is present per hepatocyte in human liver biopsies.62 cccDNA serves as a substrate of viral mRNA synthesis; it is the stable form of HBV DNA that is most resistant to antiviral treatment and the host immune response.63 In summary, the salient features of HBV replication are the use of viral RNA as a template for reverse transcription; protein-priming in which both the TP and the RT domains of the viral polymerase participate; and incomplete plus-strand synthesis that results in a partially duplex DNA genome found within the hepatitis B virion particles. HBx is a regulatory protein that is required for the viral life cycle, but its precise role is not clearly understood.64 This protein acts as a regulator of both viral and cellular gene expression. It does not directly bind DNA but appears to interact with host factors that bind DNA,65 including transcription factors and components of basal transcriptional machinery.16 The cellular targets are numerous and the list of the cellular functions HBx has been shown to modulate continues to grow.16 Overall, the functions of HBx are characterized as transcriptional transactivators. Nuclear factor kappa B (NF-κB) was one of the first HBx-responsive elements identified.66 Because the NF-κB site does not exist within the HBV genome, it is believed that the trans-activating potential of HBx extends to cellular target genes involved in inflammation. Other HBx-responsive transcription factors include ATF-2/CREB, AP-1, STAT-3, and NF-AT.67,68 Within the cytoplasm, it also localizes to the mitochondria and induces oxidative stress via calcium signaling.69,70 These activities may mediate mitochondria-mediated liver injury. HBx has also been shown to modulate cellular signal transduction pathways.71,72 It was shown to activate Src kinase and stimulate the Ras-Raf-MAP kinase signal transduction pathway.73 The physiologic significance of any of these observations remains to be established in the context of human infections. HBx is not known to be directly oncogenic, but on the basis of its role in activation of transcription factors, elevation of reactive oxygen species, and inhibition of apoptosis,74 it is considered a potential contributor to the processes of liver oncogenesis.75 HBx binds Bcl-2 proteins promoting elevation of cytosolic Ca, cell death, and viral replication.76 Envelopment of the nucleocapsid occurs in the ER or ER-Golgi intermediate compartments.16 All three forms of the surface proteins are synthesized in the ER as integral transmembrane proteins and are involved in the virion morphogenesis.77 The overall ratio of these proteins is approximately 1000 : 10 : 1. Viral assembly begins with the translation of C RNA into core and polymerase proteins. After its translation, the polymerase protein apparently binds to the ε signal on the same molecule that now functions as a pregenome, forming a preassembly complex that triggers core protein association. Core protein monomers rapidly dimerize and provide a pool of assembly intermediates. Virion formation initiates by specific interactions between core particles, and after budding, the virions are secreted via vesicular transport through the remaining compartments of the secretory pathways and are eventually released into the bloodstream. M protein is not important for morphogenesis but is required for infectivity. L protein is required for virion morphogenesis,78 and its retention in an early compartment of the secretory pathway leads to the efficiency of the assembly process. During chronic hepatitis, accumulation of the L surface protein-containing particles has been associated with the pathologic phenotype of ground-glass hepatocytes.16 The natural history of hepatitis B consists of four phases that are the result of the interplay between the virus and the host; however, it is not necessary to go through all of the phases. The first phase, the immunotolerant phase, is characterized by limited immune response against the virus, so there is minimal elevation of serum aminotransferases and minimal liver inflammation despite circulating HBsAg, HBeAg, and high levels of HBV DNA. In cases of perinatal infection, this phase may last for decades during which there are low rates of spontaneous seroconversion to anti-HBe. The cumulative rate of spontaneous HBeAg clearance during this phase is estimated to be approximately 2% during the first 3 years of life and only 15% 20 years after perinatal infection.79,80 In contrast, among immunocompetent adults, this phase is typically present only during the incubation period. During the second phase of the infection, the immunoactive phase, there is a reduction in HBV DNA levels and an increase in immune response, accompanied by increased aminotransferases and liver inflammation. This is thought to be the period in which there is augmentation of both innate and acquired HBV immunity, leading to cytolytic destruction of hepatocytes, as the peak of the immune response coincides with the aminotransferase elevations.81 In adults, this is usually the period of symptomatic acute hepatitis B, which can last for decades if the immune reaction is not sufficiently vigorous to lead to viral clearance. The third phase heralds the conversion from HBe antigenemia to anti-HBe, which can be accompanied by an increase in aminotransferases above baseline, suggesting that there is greater immune control of the virus than in the second phase.82 Because IgM anti-HBc may increase during these flares, this may lead to the erroneous diagnosis of acute hepatitis. In perinatally acquired infection, this period usually occurs in the second to third decade of life, and seroconversion rates may approach 10% to 20% per year.79,83 Similar rates are observed in adults with chronic infection. In a study of 1536 Alaskan natives who acquired HBV as adults, two thirds of subjects cleared HBeAg and developed anti-HBe during 12 years of follow-up without reversion to HBeAg positive state.84 The likelihood of seroconversion is inversely related to the serum aminotransferase. Spontaneous seroconversion occurs in 50% of those with serum aminotransferases greater than five times the upper limit of normal compared with less than 10% in those with lower aminotransferases.85 Spontaneous seroconversion is marked by the appearance of HBcAg-specific CD4+ and CD8+ T cells,86 but the precise factors preceding this event are unknown, and seroconversion cannot be linked to specific viral mutants, as was previously believed.87 Males and older individuals are also more likely to have spontaneous seroconversion, and viral genotype may play a role.40 During this third phase, one of three outcomes occurs. The most common is a decrease in viral replication and reduction in aminotransferases with low or undetectable HBV DNA and is referred to as inactive hepatitis B. Termination of virus replication is associated in most patients with biochemical and histologic regression of inflammatory activity.88 Some individuals will also clear HBsAg, although this is unusual, occurring in less than 1% of patients per year in adults and 0.05% to 0.8% in infection acquired in infancy or childhood.84,89 The second outcome is seroreversion to HBeAg positivity and return to the immunoactive phase, which occurs 10% to 40% of the time. The third outcome is that patients remain with high levels of HBV DNA and elevations of ALT but remain HBeAg negative, which occurs about 20% of the time. The fourth phase is the resolution phase in which HBsAg clearance occurs and anti-HBs develops. It has been difficult to understand the mechanism for viral persistence and immune-mediated liver injury because research in this area is hampered by the lack of a robust culture system or a small animal model. Most studies suggest that HBV is not directly cytopathic to the hepatocyte,90,91 which is also supported by the existence of asymptomatic hepatitis B carriers with normal liver histology and function. Both human and animal studies have now shown that the liver injury mediated by HBV is initiated by a viral-specific cellular and humoral immune response. In more than 95% of immunocompetent adults, the immune response is vigorous, polyclonal, and multispecific and results in acute, self-limited hepatitis with reduction of viral load and the development of long-lasting humoral and cellular immunity. Persistent infection is associated with necroinflammatory activity, which eventually leads to cirrhosis. Natural recovery from acute hepatitis B probably depends on multiple components of cellular immune responses, including natural killer (NK) cells, natural killer T (NK-T) cells, and virus-specific CD4+ T cells and CD8+ cytotoxic T lymphocytes (CTL). However, a hallmark of acute hepatitis B is failure to induce an innate cytokine response mediated by pattern recognition receptors.92,93In self-limited infections, the noncytopathic response leads to lowering of the HBV DNA by more than 90% within a few weeks of peak HBV replication and before liver damage occurs.94 Both NK and NK-T cells also contribute to clearance through production of IFN-α/β.95 Acute HBV infection is also accompanied by a strong and transient expansion of CD4+ T cells directed against multiple epitopes within the HBV. HBc is the dominant antigen recognized by CD4+ T cells in most cases of acute, resolving HBV infection.95a These HBc-specific CD4+ T cells provide help for the production of antibody to HBsAg and are also associated with the development of a vigorous and polyclonal cytotoxic CTL capable of recognizing different epitopes within the HBV genome.96 Individuals who successfully clear HBV infection, either spontaneously or after IFN therapy, maintain these broad and strong peripheral CTL responses.97 CD4+ and CTL memory in the presence of low levels of persisting HBV DNA has been shown to persist up to 23 years after infection despite markers of serologic recovery. Both virus-specific CD4+ and CTL contribute to control of HBV replication through both direct cytolysis of infected cells but, more importantly, through production of cytokines that control viral replication.98 Analysis of the NK, CTL, and CD4+ T-cell responses in the incubation phase has demonstrated that CTL response increases in parallel with alanine aminotransferase (ALT), consistent with the previously observed notions that CTL activity is responsible for liver injury, and peaks about the time that the HBV DNA titers begin to fall.99 Recently, regulatory T cells were found to influence the immune response during acute HBV infection by decreasing cytokine production of effector T cells, thereby downregulating the antiviral activity. This limits the liver injury during acute infection but as a consequence prolongs viral clearance.100 The development of surface antibody (anti-HBs) follows the disappearance of HBsAg and marks recovery from acute hepatitis B, which occurs in 95% of adults after acute infection. Anti-HBs is sufficient for protection against HBV infection, as demonstrated by the success of the current HBV vaccines, even if it is not the sole operative mechanism clearing acute infection. The “a” determinant is the predominant B-cell epitope common to all HBV subtypes. Antibodies against this epitope confer immunity to all HBV subtypes. Coexistence of HBsAg and anti-HBs is reported in up to 24% of chronically infected individuals, in which case the anti-HBs is directed against one of the subtypic determinants and a mutation preventing neutralization of the virus has occurred. Other surface antigens that stimulate antibody responses include the pre-S1 and pre-S2 antigens. Antibody to these develops during recovery and can be detected before anti-HBs; however, routine serologic assays are not readily available. Mutation in the S gene usually occurs in the “a” determinant, which allows the virus to escape antibody neutralization. This may occur following the use of hepatitis B immune globulin (HBIG) for passive immunization of newborns and transplant recipients. Persistent HBV infection may result because of the failure of initial innate and adaptive immune responses. Among infants who become infected at birth, both viral and host factors play a role in the development of chronic infection. The presence of HBeAg and the viral titer in the mother are both directly related to the likelihood of infant infection. In animal models, HBeAg may be tolerogenic,101 and because HBeAg and HBcAg are cross-reactive at the T-cell level, deletion of the CD4+ HBc-specific T-cell responses results in ineffective CTL responses to HBcAg. Adult immunocompetent individuals who fail to clear acute HBV infection also have less vigorous CD4+ T-cell and CTL responses. In contrast to acute resolving infection, peripheral CD4+ T-cell and CTL responses in those individuals who develop chronic infection are weak and more narrowly focused.102 The mechanisms by which HBV evades the immune response and results in chronic infection in these adults remain obscure. In part this is determined by host genetic factors because persons with certain human leukocyte antigen (HLA) alleles appear to be more susceptible to chronic infection,103–106 and certain HLA-DP alleles have been associated with recovery from acute HBV infection.107,108 It has also been suggested that the size of initial viral inoculum and viral kinetics may be such that the immune system is overwhelmed by the virus and becomes “exhausted.”109 Despite the weak and narrowly focused response of the peripheral blood CTL, HBV-specific CD4+ and CD8+ T cells can be isolated from the livers of chronic hepatitis B patients that are capable of ex vivo, class-I-restricted cytolytic activity in response to envelope and core peptides. The cytotoxicity mediated by these cells appears to be sufficiently strong to cause liver injury but not strong enough to eradicate virus from all hepatocytes.110,111 In addition to the virus-specific cells, the inflamed liver contains other cells that may participate in hepatocyte damage.112
Hepatitis B Virus and Hepatitis Delta Virus
Introduction
Historical Background and Classification
Biology
Attachment, Entry, and Hepatotropism
Viral Genome
Transcription
Translation
Replication
HBx
Morphogenesis and Assembly
Natural History and Pathogenesis of Disease
Natural History
Pathogenesis
Acute Hepatitis B
Chronic Hepatitis B
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Hepatitis B Virus and Hepatitis Delta Virus
148