COMMON GENETIC ALTERATIONS IN HEAD AND NECK SQUAMOUS CELL CARCINOMA
P16/p53/Cyclin D
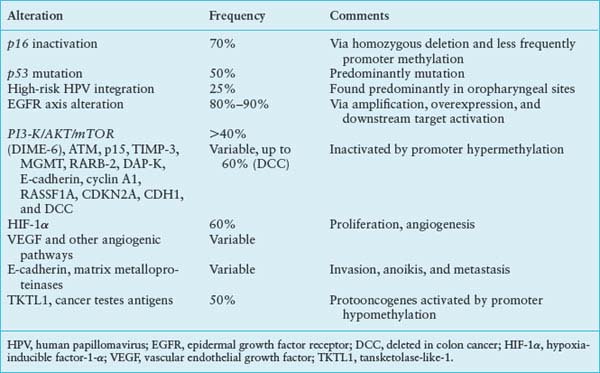
Loss of 9p21, resulting in inactivation of the p16 gene, is the most common genetic alteration in the progression of head and neck cancer.19,20 P16 is an inhibitor of cyclin-dependent kinase (CDK), which is intimately involved in G1 cell-cycle regulation. Phosphorylation and inactivation of pRb by unbridled CDK4 and CDK6 enable cells to escape senescence. Loss of chromosome 9p21 occurs in the majority of invasive tumors and is also present at a high frequency in the earliest definable lesions, including dysplasia and carcinoma in situ.19 Loss of p16 appears necessary for immortalization of keratinocytes.21 Loss of p16 protein has been observed in most advanced premalignant lesions.22 In addition to deletions and point mutations, p16 is also inactivated by methylation of the 5’CpG region.23 This methylation is associated with complete block of p16 transcription and appears to be a common mechanism for p16 inactivation. The notion that p16 inactivation is directly involved in the progression of primary tumors has been strengthened. Lack of p16 protein was detected by immunostaining in most primary invasive lesions, and tumors with absent p16 protein contained a homozygous deletion, methylation, or point mutation of p16.24
Loss of p53 on chromosomal region 17p13 and subsequent point mutation within the remaining allele is another critical step in tumor progression. Inactivation of p53 now represents the best-described and most common genetic change in all of human cancer.25 Initially by analysis of exons 5-8, p53 mutations were observed in approximately 50% of head and neck tumors.26,27 The p53 gene can be inactivated by a large variety of distinct mutations and more thorough sequence analyses of exons 2-11, a p53 mutation rate of almost 80% has been observed in head and neck tumors.28 p53 normally halts cell-cycle progression in the setting of DNA damage and induces apoptosis with inadequate DNA repair. p53 mutations result in a progression from preinvasive to invasive lesions, while increasing the probability of further progression. If 17p loss or p53 mutation is present in early lesions, the chance of progression to cancer within 10 years approaches 80% (33-fold relative risk). In a large definitive collaborative group study, disruptive p53 mutations were an independent prognostic marker and predicted a worse outcome in surgically resected primary tumors.29
Carcinogens in tobacco and alcohol have a causal role as the prevalence of p53 mutations is greater in patients who smoke and drink alcohol.30 HPV16- and HPV18-induced SCC of the oropharynx is more common in nonsmokers and is not associated with p53 mutations. Instead, the viral oncoprotein E6 promotes the accelerated, ubiquitin-mediated, degradation of p53.
Amplification of chromosome region 11q13 containing cyclin D1 is seen in approximately one-third of head and neck tumors.31 The role of cyclin D1 in the progression of human cancer is now well established and constitutive activation of oncogene cyclin D1 has been shown to confer a growth advantage in SCCs.32 Other tumor suppressor genes, including Rb and p16, are negative regulators of the cyclin D1 pathway and often are inactivated in human neoplasms. Cyclin D1 amplification is independent of p16 inactivation in head and neck cancers.33
PI3-K/AKT/mTOR
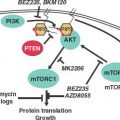
Mutation in the PI3-K signaling pathway are found in up to 30% of all human cancers, and activation of the PI3-K/AKT/mTOR pathway has also been implicated in tumorigenesis of HNSCC.34 Mutations of the PI3-K network have shown to confer a growth advantage, transforming capacity, and drug resistance.35 The PI3-K family is divided into three different classes based on structure and substrate specificity. Class I PI3-Ks are heterodimers of a p85 regulatory subunit and a p110 catalytic subunit, which is mutated in many cancers. The class I PI3-Ks are activated by tyrosine kinase receptors, including epidermal growth factor receptor (EGFR) and oncogenic proteins, and lead to the production of the lipid second messenger, phosphatidylinositol 3-phosphatase (PIP3), which in turn facilitates phosphorylation and subsequent activation of AKT. The PI3-K pathway also leads to activation of the serine/threonine kinase, mammalian target of rapamycin (mTOR), which in turn phosphorylates p70S6K, a kinase that modulates protein synthesis.
Invasion/Metastasis
Metastasis is a complicated, multistep process in which selective pressures select for a clone of malignant cells selected to survive in a distant, permissive environment. The multistep process includes angiogenesis, altered cellular adhesion, cellular motility, disruption of the base membrane/extracellular matrix, and anchorage-independent proliferation.
Up-regulation of hypoxia-inducible factor 1-α (HIF-1α), induced by intratumor hypoxia, has been documented in invasive HNSCC and correlates with progression to a more invasive and aggressive phenotype.36,37 HIF-1α is a master regulator of oxygen homeostasis and activates genes involved in angiogenesis, glucose metabolism, cell survival, invasion, cell renewal, and immortalization.38–40
The vascular endothelial growth factor (VEGF) pathway is critical in angiogenesis in HNSCC. Increased expression of VEGF and its receptors is regulated by HIF-1α–dependent and -independent pathways, both of which converge on the PI3-K/AKT pathway.
Diminished cell-to-cell adhesion, through down-regulation of cellular adhesions molecules such as E-cadherin, is integral to invasion.41,42 In addition to cell-to-cell adhesion mediated by cadherins, integrins that mediate cell-to-extracellular matrix interaction also play a fundamental role in tumor cells gaining access to the angiolymphatic system.43 Laminins, which are extracellular glycoproteins, are one of the ligands for integrins. Laminin 5 is overexpressed in invasive fronts, is associated with poorer prognosis, and downstream activates mitogen-activated protein kinase, which leads to cell survival and proliferation.44–51 In addition to altered cell adhesion, migration mediated by the Rho family of GTPases is an important step in the multistep process of metastasis.52 Cancer cells also actively disrupt the base membrane through the proteolytic activity of zinc-dependent endopeptidases, matrix metalloproteinases (MMPs), to disseminate. MMPs degrade most components of the base membrane, including collagen. Evasion from anchorage-dependent survival is a feature of metastatic HNSCC, and anoikis resistance is crucial in the process of metastasis as a defense against microenvironmental death stimuli. E-cadherin has been implicated in conferring anoikis resistance in HNSCC by physically associating with a number of signaling effectors such as PI3K and EGFR.53,54
Epidermal Growth Factor Receptor
The EGFR is one of the best-studied oncogenes in HNSCC. This receptor tyrosine kinase belongs to the ErbB family of cell surface receptors and has many downstream signaling targets associated with carcinogenesis. Once phosphorylated, the receptor can signal via the MAPK, Akt, ERK, and Jak/STAT pathways (Fig. 14.1). These pathways are related to cellular proliferation, apoptosis, invasion, angiogenesis, and metastasis.55–57 Expression of EGFR is a normal finding in many tissues including the dermis, gastrointestinal tract, and kidneys. However, dysfunction of this receptor and its associated pathways occurs in most epithelial cancers55 and 80% to 90% of HNSCC specifically.9,56 The story of EGFR is promising in that our understanding of its molecular biology has led directly to clinically beneficial targeted therapies and its use as a marker and prognosticator of disease.
Initially, EGFR was first found to be up-regulated in HNSCC cell lines and in a high percentage of primary HNSCC.58–60 Further study showed that histopathologically normal mucosa adjacent to cancer had a high degree of overexpression61 and that the up-regulation of EGFR occurs in the transition from dysplasia to HNSCC.62 Now it is well known that elevated levels of expression predict a worse disease-free and cause-specific survival.63 Studies looking at copy number amplification have also been shown to be associated with poorer prognosis in HNSCC. One study has demonstrated that the overexpression of EGFR is a biomarker for an improved response to therapy and could serve as a predictive marker to separate patients into different arms of therapeutic trials.64
In addition to serving as a marker for prognosis, EGFR axis alterations are currently under investigation as markers for response to treatment and as therapeutic targets. Several strategies exist for targeting the EGFR pathway including the use of specific tyrosine kinase inhibitors (TKIs), monoclonal antibodies blocking receptor dimerization, and antisense oligodeoxynucleotides or siRNA blocking mRNA expression.
Cetuximab is one of the most well studied monoclonal antibodies directed against EGFR. A recently published phase 3 clinical trial examined the effects of this drug in conjunction with radiotherapy in the treatment of locoregionally advanced HNSCC. This study demonstrated an overall survival benefit (49 vs. 29 months) and increased duration of locoregional control (24.4 vs. 14.9 months) in the cetuximab plus radiotherapy arm versus the arm receiving radiotherapy alone. This was the first randomized study showing a survival benefit with an EGFR targeting agent in locally advanced HNSCC.65,66 Conversely, the TKI gefitinib has shown no survival benefit for recurrent or metastatic HNSCC.66,67 There are, however, several phase 1 and 2 trials under way studying the concomitant use of chemoradiation with gefitinib and two other TKIs (erlotinib and lapatinib) in the treatment of HNSCC.68 There are several other studies currently investigating the role of EGFR targeting agents, three of which are mentioned here. The first trial is evaluating gefitinib plus docetaxel versus placebo on recurrent or metastatic disease. The second trial is examining the role of cetuximab as an adjuvant to cisplatin and 5-fluorouracil therapy in recurrent/metastatic disease. The last notable trial is studying cetuximab as an adjuvant to radiotherapy and cisplatin treatment for locally advanced disease.66 A shortcoming of all these clinical trials is that they fail to incorporate the presence of EGFR-activating mutations, EGFR gene copy number, or both, into their primary analyses, although both of these factors have recently emerged as predictors of efficacy.66 However, retrospective analysis of these factors within the context of prospective therapeutic trials should provide some information.
As mentioned previously, increased EGFR expression correlates with a poor prognosis; conversely, the presence of activating somatic mutations in the EGFR-TK domain have been shown to be a positive predictor for a patient’s response to treatment. These mutations, which are present in ∼10% of NSCLC69,70 and 1% to 7% in non– small cell lung cancer (HNSCC),71,72 correlated with prolonged survival in patients with advanced NSCLCs treated with chemotherapy regardless of EGFR-TKI.73 Several other studies in NSCLCs did show that EGFR mutations were predictive of survival in patients treated with EGFR-TKIs.74–76 Multiple clinical trials are under way to evaluate the use of EGFR-TKIs in chemotherapy-naive patients with advanced NSCLC and EGFR-activating mutations.66,77,78 Both gefitinib and erlotinib have demonstrated response rates of 75% to 82%.66,77,78 It is yet to be determined whether the same correlation and response to therapy will be found in HNSCC with EGFR-activating mutations.
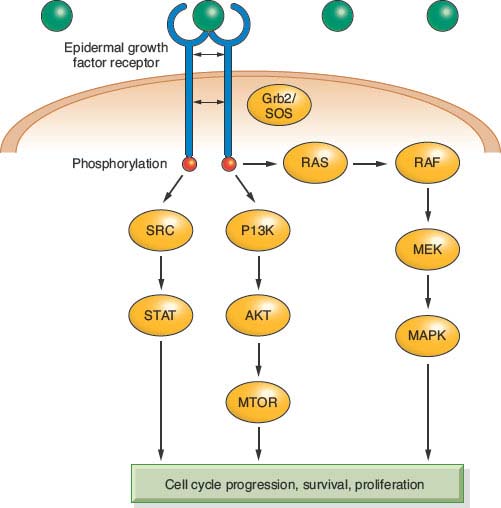
FIGURE 14.1 Cell-cycle progression, survival, and proliferation.
Human Papillomavirus in HNSCC
Although it is well known that tobacco and alcohol are the two primary environmental risk factors associated with the development of HNSCC, it is now recognized that HPV infection plays an important role in the pathogenesis of a unique subset of oropharyngeal HNSCCs.79–85 These tumors primarily emerge from the lingual and palatine tonsils in the oropharynx.80,83,86–91 HPV-related HNSCC has distinguished itself as a separate entity with an improved prognosis and response to therapy from non-HPV–related HNSCC. It has also been proposed that it is the reason for the increasing incidence of oropharyngeal HNSCC relative to all other anatomic sites in the head and neck. This section will first briefly review what is known about the molecular biology of HPV as this topic is covered in other chapters within this edition, and then move on to how HPV-related HNSCC affects everyday clinical practice.
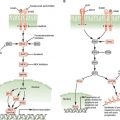
HPV is an ∼7.9-kb, nonenveloped, double-stranded, circular DNA virus that has a specific tropism for squamous epithelium.92 Although the sequences for over 320 different types of HPV have been identified, HPV-16 and -18 are the two types most pertinent to the development of HNSCC. The E6 and E7 oncoproteins contained within the viral genome, if overexpressed, are able to disrupt the function of Rb and p53, well-known tumor suppressor genes, leading to development of a malignant phenotype (Fig. 14.2).93 It is now established that histopathologically, HPV-positive tumors tend to have a poorly differentiated and frequently basaloid histology that often lacks keratin.79,80,85,94 Researchers have also discovered that human keratinocytes expressing E6 and E7 genes from HPV-16 become immortal,95 as do oral epithelial cells.96–98
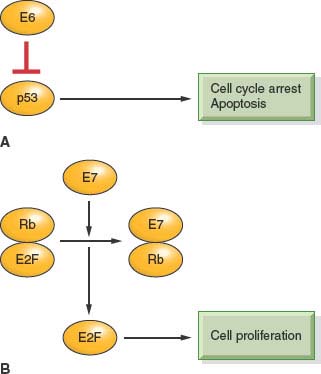
FIGURE 14.2 A: E6 binds p53, targets it for degradation, and promotes cell-cycle progression. B: E7 binds Rb and releases E2F, which promotes cellular proliferation.
In a recent meta-analysis, HPV genomic DNA was detected in approximately 26% of all HNSCC by sensitive polymerase chain reaction–based methods.99 However, in the majority of studies, 50% or more of oropharyngeal tumors contained the HPV genome.100 A multinational study conducted by the International Agency for Research on Cancer (IARC), only 18% of oropharyngeal tumors were HPV-positive, indicating that this proportion likely varies by geography.101 Regardless of the study population, high-risk HPV-16 accounts for the overwhelming majority (90% to 95%) of HPV-positive tumors.99
Our knowledge of HPV and its causal relationship with oropharyngeal cancer has improved our ability to diagnose and locate disease in patients with occult primary tumors.102 Tonsillectomy has been shown in retrospective analyses to identify the primary site of cervical metastases as the contralateral or ipsilateral tonsil in approximately 10% and 30% of cases, respectively.103–106 Therefore, HPV-related cancer is a distinct established entity that can be reliably diagnosed.
On average, patients with HPV-positive HNSCC are approximately 5 years younger than HPV-negative HNSCC patients, with equal distribution among the sexes.90,107–110 HPV-positive HNSCC is more likely than HPV-negative HNSCC to occur in the nonsmoker and nondrinker.80,81,88,89 Risk factors for HPV-related HNSCC include a high lifetime number of vaginal-sex partners (26 or more), a high lifetime number of oral-sex partners (6 or more),111 and seropositivity for HPV-16 viral capsid protein antibodies,82 which carries a 15-fold increased risk for HNSCC. In a study restricted to patients with oropharyngeal cancers, nonsmokers were approximately 15-fold more likely to have a diagnosis of HPV-positive HNSCC than smokers.112
Our knowledge of the HPV status of our patients’ tumors also improves our ability to provide an accurate prognosis. Patients with HPV-positive tumors have improved prognosis when compared with patients with HPV-negative tumors in the majority of studies, with as much as 60% to 80% reduction in risk of dying from their cancer when compared with the HPV-negative patient after controlling for other risk factors.80,108,112–115 The reason for the improved survival is unclear; however, improved radiation responsiveness, immune surveillance to viral antigens, and the absence of field cancerization in these patients who tend to be nonsmokers have been postulated as possibilities. In addition, E6-related degradation of p53 in HPV-positive cancers may not be functionally equivalent to HPV-negative p53 mutations, and therefore HPV-positive tumors may have an intact apoptotic response to radiation and chemotherapy.
Therapeutic implications of an HPV-positive diagnosis are an active area of investigation. The Eastern Cooperative Oncology Group is studying the impact of HPV presence on oropharyngeal organ-preservation therapy. It is believed that HPV-HNSCC will perform better than HPV-negative HNSCC. A clinical trial of an HPV-16–specific therapeutic vaccine is also currently being evaluated. The vaccine is administered in the adjuvant setting and is intended to enhance the cytotoxic T-cell response to the HPV-16 oncoproteins.116 With regard to prevention, a prophylactic vaccine composed of the HPV-16 viral capsid protein has recently been shown to prevent persistent HPV-16 infection and the development of cervical dysplasia.117,118 However, the clinical trials have not included an evaluation of the impact of the vaccine on oral HPV infection. The vaccine does have the potential to have an impact on HNSCC incidence because the current vaccines are targeted to HPV-16 and there are animal models that demonstrate a protective effect and a reduction in the development of HPV-related oral lesions.
Possible future diagnostic tests that would likely have high specificity but low sensitivity for a diagnosis of HPV-associated HNSCC will include the detection of HPV-16 DNA in plasma,119 which can be used for surveillance. Other screening tests like fluorescence in situ hybridization (FISH) on Papanicolaou smears obtained directly from tumors and HPV-16 E6 and E7 seroreactivity are other tests currently being tested.
EPIGENETICS
Regulation of gene expression by DNA methylation was first recognized in development, where the coordinated expression and silencing of genes needs to take place in an organized fashion.120 As a novel mechanism of gene regulation, it was quickly proposed that epigenetic control of tumor suppressor genes could be an important mechanism of carcinogenesis.121,122 To date, methylation has been primarily considered as a mechanism of tumor suppressor gene inactivation, and one of the earliest genes to be characterized as being epigenetically controlled was the retinoblastoma gene. Primary tissue analysis showed that 10% of the retinoblastomas analyzed were hypermethylated at the Rb promoter123 in the absence of any other mutations. In recent years, new assays such as sodium bisulfite treatment of DNA, which converts nonmethylated cytosines to uracil and methylation-sensitive quantitative polymerase chain reaction, have further advanced our ability to evaluate the methylation status of tissue samples.124–126 With these advances, many different tumor suppressor genes in various tumor types have been shown to be down-regulated by methylation. The utilization of comprehensive whole-genome profiling approaches to promoter hypermethylation has identified novel putative tumor suppressor genes silenced by promoter hypermethylation. These in vitro techniques employ treatment of cultured cells with pharmacologic demethylating agents and subsequent expression array analysis with validation of tumor suppressor gene targets.127
Promoter hypermethylation of p16 is a frequent event in HNSCC and this mechanism of gene silencing accounts for the low levels of expression.23,24,123,128,129 Thus, Knudsen’s two-hit hypothesis could include promoter hypermethylation as one of the “hits” along with the more traditional sequence mutation or chromosomal deletion.
Studies of promoter methylation have uncovered many other putative tumor suppressor genes in HNSCC including lhx-6 (DIME-6), ATM, p15, TIMP-3, MGMT, RARB-2, DAP-K, E-cadherin, cyclin A1, RASSF1A, CDKN2A, CDH1, and DCC. These genes are known to function in pathways that control cell-cycle progression, apoptosis, cell-cell adhesion, DNA repair, and tumor invasion.130–141 With the advent of new molecular techniques and whole-genome screening strategies, the list of tumor suppressor genes that are silenced through promoter hypermethylation continues to grow at a rapid pace. For instance, in the relatively young field of microRNA research, it has been demonstrated that those microRNAs having tumor-suppressor function also undergo DNA methylation-associated silencing in cancer.137,142,143
LOSS OF HETEROZYGOSITY AND RISK OF MALIGNANT PROGRESSION
Premalignant lesions of the head and neck are often characterized by large patches of clonally related precursors, often demonstrating dysplastic changes. Increased risk of progression to malignancy is also associated with prior head and neck cancer, advanced histologic grade, and evidence of genetic instability including chromosomal loss and aneuploidy. Those patients with moderate or severe dysplasia are noted to have a risk of progression to malignancy of approximately 60%, in a study with median 7-year follow up. Likewise, a prior history of head and neck cancer resulted in a similar 60% risk of progression in the same study.6 Approximately 40% of patients with mild dysplasia or hyperplasia combined with 3p or 9p loss of heterozygosity demonstrated progression to malignancy within 5 years.
Selected References
The full list of references for this chapter appears in the online version.
1. Foulkes WD, Brunet JS, Sieh W, Black MJ, Shenouda G, Narod SA. Familial risks of squamous cell carcinoma of the head and neck: retrospective case-control study. BMJ 1996;313:716.
6. van Zeeburg HJ, Snijders PJ, Wu T, et al. Clinical and molecular characteristics of squamous cell carcinomas from Fanconi anemia patients. J Natl Cancer Inst 2008; 100:1649.
12. Califano J, van der Riet P, Westra W, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res 1996;56:2488.
16. Mao L, Lee JS, Fan YH, et al. Frequent microsatellite alterations at chromosomes 9p21 and 3p14 in oral premalignant lesions and their value in cancer risk assessment. Nat Med 1996;2:682.
23. Reed AL, Califano J, Cairns P, et al. High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res 1996;56:3630.
28. Poeta ML, Manola J, Goldwasser MA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med 2007;357:2552.
36. Hoogsteen IJ, Marres HA, Bussink J, van der Kogel AJ, Kaanders JH. Tumor microenvironment in head and neck squamous cell carcinomas: predictive value and clinical relevance of hypoxic markers. A review. Head Neck 2007;29:591.
41. Mandal M, Myers JN, Lippman SM, et al. Epithelial to mesenchymal transition in head and neck squamous carcinoma: association of Src activation with E-cadherin down-regulation, vimentin expression, and aggressive tumor features. Cancer 2008;112:2088.
55. Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol 2006;24:2666.
64. Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med 2006;354:567.
78. Andl T, Kahn T, Pfuhl A, et al. Etiological involvement of oncogenic human papillomavirus in tonsillar squamous cell carcinomas lacking retinoblastoma cell cycle control. Cancer Res 1998;58:5.
79. Gillison ML, Koch WM, Capone RB, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst 2000; 92:709.
80. Hafkamp HC, Speel EJ, Haesevoets A, et al. A subset of head and neck squamous cell carcinomas exhibits integration of HPV 16/18 DNA and overexpression of p16INK4A and p53 in the absence of mutations in p53 exons 5-8. Int J Cancer 2003;107:394.
81. Mork J, Lie AK, Glattre E, et al. Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. N Engl J Med 2001;344:1125.
82. Schwartz SM, Daling JR, Doody DR, et al. Oral cancer risk in relation to sexual history and evidence of human papillomavirus infection. J Natl Cancer Inst 1998;90:1626.
110. D’Souza G, Kreimer AR, Viscidi R, et al. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med 2007;356:1944.
120. Feinberg AP, Vogelstein B. Hypomethylation of ras oncogenes in primary human cancers. Biochem Biophys Res Commun 1983;111:47.
127. El-Naggar AK, Lai S, Clayman G, et al. Methylation, a major mechanism of p16/CDKN2 gene inactivation in head and neck squamous carcinoma. Am J Pathol 1997; 151:1767.
140. Ha PK, Califano JA. Promoter methylation and inactivation of tumour-suppressor genes in oral squamous-cell carcinoma. Lancet Oncol 2006;7:77.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


