FIGURE 23.1 Tissue-restricted genetic instability in ovarian epithelial cells from women at risk for ovarian cancer with no BRCA mutations. Mitomycin C–induced chromosomal breakage is high and FANCD2 levels are low in ovarian epithelial cells but normal in peripheral blood lymphocytes and may antedate the onset of overt carcinoma. (We thank Dr. Grover Bagby for helping to create this figure.)
Interestingly, tumors with inactivated BRCA2 are responsive to cisplatin. However, due to their low accuracy of DNA repair, these cells accumulate secondary genetic modifications that can lead to reversal of BRCA2 mutation, allowing these cells to acquire resistance to crosslinking agents.10
Other DNA Repair Pathways
Similar to the FA/BRCA pathway, disruptions of other DNA repair pathways have been observed in ovarian cancer. These disruptions account, at least in part, for the specific drug sensitivity of the tumors. Recent studies indicate translesion DNA synthesis defect in ovarian cancer is a consequence of elevation in activity of POLB, an error-prone polymerase. Inhibition of POLB in these cells results in resensitization to cisplatin.11 Overall it is believed that although inactivation of one DNA repair pathway may confer advantage to tumors, cancer cells may rely more on other repair pathways. Therefore, inactivation of the second pathway would be deleterious for these cells, causing synthetic lethality. An RNA interference screen identified the ataxia-telangiectasia mutated (ATM) pathway to be synthetically lethal with FA.12 Similarly a strategy for synthetic lethality is under investigation, using base excision repair poly(adenosine diphosphate-ribose) polymerase 1 (PARP1) inhibitors in the treatment of homologous recombination deficient ovarian cancer.13,14 PARP inhibition has been shown to be up to 1,000 times selectively more toxic to cancer cells than to wild type cells. PARP inhibitors act by exploiting a tumor cell’s defect in homologous recombination, a type of DNA repair. This is because following PARP inhibition, cells require homologous recombination to repair common types of DNA damage. Although normal cells can use homologous recombination for repair of this damage and survive, certain types of tumors (e.g., those with BRCA1 or BRCA2 defects) have lost the ability to repair by homologous recombination and will die.
Genome Wide Association Studies. The identification of common ovarian cancer susceptibility variants may have clinical implications in the future for identifying patients at greatest risk of the disease. In this regard, several genome wide association study (GWAS) have been performed in ovarian cancer. The most striking of these was a recent study by the Ovarian Cancer Association Consortium to identify common ovarian cancer susceptibility alleles.15 A total of 507,094 single nucleotide polymorphisms (SNPs) were genotyped in 1,817 cases and 2,353 controls from the United Kingdom; and 22,790 top ranked SNPs were also genotyped in 4,274 cases and 4,809 controls of European ancestry from Europe, the United States, and Australia. Twelve SNPs were identified at 9p22 associated with disease risk (P <10−8). The most significant SNP (rs3814113; P = 2.5 × 10−17) was genotyped in a further 2,670 ovarian cancer cases and 4,668 controls, confirming its association (combined data odds ratio [OR] = 0.82 95% confidence interval [CI], 0.79 to 0.86; Ptrend = 5.1 × 10−19). The association was strongest for serous ovarian cancers (OR 0.77; 95% CI, 0.73 to 0.81; Ptrend = 4.1 × 10−21).
Transcriptional Profiling of Ovarian Cancer Histologic Subtypes
Several gene expression studies using cDNA microarrays have been performed in ovarian cancer. Additionally, several studies have focused on the alterations demonstrated in the DNA copy number.16–18 Array-based technology has shown that the different histological subtypes of ovarian carcinoma are distinguishable based on their overall genetic expression profiles. A common finding among several studies is the ability to distinguish low-grade serous ovarian carcinoma from high-grade carcinoma based on their gene expression profiles.17,19–23 A number of genes shown to be differentially expressed in EOC are known to be involved in many important cellular mechanisms, including cell cycle regulation, apoptosis, tumor invasion, and control of local immunity.20,24,25
Increased mutagenic signaling by receptor tyrosine kinases plays a major role in ovarian carcinogenesis (Table 23.1). Overexpression of epidermal growth factor receptor (EGFR) (ERBB1), ERBB2/HER2/neu, and c-FMS has been reported repeatedly in ovarian cancer. One of the major downstream mediators of signaling initiated by these receptors is the phosphatidylinositol 3-kinase (PI3K)–AKT pathway. Aberrations in this pathway including increased AKT1 kinase activity, AKT2 and PI3K amplification, and PI3KR1 mutations may provide opportunities for therapeutic intervention.
TABLE 23.1
GENETIC ALTERATIONS IN OVARIAN CANCER
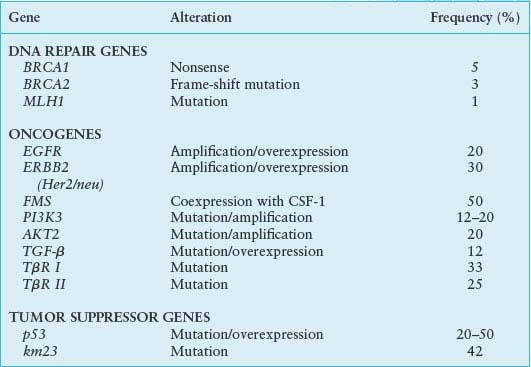
It has been reported that more than 75% of ovarian carcinomas are resistant to transforming growth factor-beta (TGF-β),26 and the loss of TGF-β responsiveness may play an important role in the pathogenesis or progression of ovarian cancer. In addition, it has been shown that TGF-β1, the TGF-β receptors (TβR-II and TβR-I), and the TGF-β signaling component Smad2 are altered in ovarian cancer. Alterations in TβR-II have been identified in 25% of ovarian carcinomas, whereas mutations in TβR-I were reported in 33% of such cancers.27 Protooncogene transformation might lead either to an overexpression of mitogenic molecules or an inactivation of those with inhibitory action, thus contributing to neoplastic transformation and development. The most important protooncogenes of the first group are undoubtedly constituted by FMS and HER2/neu. The first one encodes a transmembrane tyrosine kinase receptor, which binds MCSF. It is possible that FMS-MCSF both stimulates epithelial cell proliferation and induces a chemical attraction for macrophages that, in turn, can produce mitogenic stimulating factor. Elevated plasma concentrations of MCSF are present in the sera of 70% of patients with ovarian cancer.28 The second protooncogene, HER2/neu, encodes another tyrosine kinase, which is similar to EGFR. Its action may consist of amplification of mitogenic action in target cells; this oncogene is overexpressed in 30% to 35% of ovarian cancer and is associated with a poor prognosis.29
Metastasis of Ovarian Cancers
Metastasis is the functional hallmark of all cancer. In general, metastasis involves the invasion of transformed epithelial cells across their basement membrane, through the underlying stroma, and into blood vessels and lymphatic channels, which subsequent disseminate them to distant sites. Only a tiny fraction of cells released into circulation by a tumor ever results in metastasis; understanding the mechanisms by which those cells can land and grow is a priority for cancer researchers. Given the unique need to accommodate the survival of exfoliated cells as well as their subsequent attachment and growth, it seems reasonable to assume that expression and functional organization of molecular pathways important for promoting the metastasis of ovarian cancers will differ from breast and other cancers that depend on hematogenous or lymphatic dissemination. Nonetheless, a wide variety of gene products implicated in the metastasis of other cancers have also been implicated in the metastasis of ovarian cancer. These include growth factor receptors such as EGFR, insulinlike growth factor receptors (IGFRs), and kinases, such as jak/stat, focal adhesion kinase, PI3K, and c-met. Comparisons of primary and metastatic ovarian cancers by transcriptional profiling have failed to reveal significant differences in the expression of gene products likely related to the metastatic process.
Particular attention has recently focused on the role of lysophosphatidic acid (LPA) in promoting the metastasis of ovarian cancers. LPA is constitutively produced by mesothelial cells lining the peritoneal cavity; its levels are increased in the ascites of women with both early- and late-stage ovarian cancers.30 When applied to ovarian cancer cell lines in vitro, LPA promotes both the migration of these cells in a manner dependent on Ras MEK kinase-1 as well as their invasion across artificial barriers analogous to a basement membrane. At a molecular level, exogenous LPA enhances ovarian cancer invasiveness both by activating matrix metalloproteinase-2 via membrane-type-1-matrix metalloproteinase (MT1-MMP) and down-regulating the expression of specific tissue inhibitors of metalloproteinases (TIMP-2 and -3).31 Its application to cultured ovarian cancer cells has also been shown to promote disassembly of intracellular stress fibers and focal adhesions,32 observations consistent with the idea that LPA promotes dissemination of ovarian cancer by loss of cell adhesion. However, LPA has also been shown to promote the invasiveness of ovarian cancers by additional mechanisms dependent on interleukin-8. The G12/13-RhoA and cyclooxygenase pathways have also been implicated in the LPA-induced migration of ovarian cancers. These mechanisms appear to be independent of the ability of LPA to induce changes in MMP2 expression. Lastly, it should be noted that LPA appears to promote ovarian cancer metastasis by stimulating fas-ligand expression and the shedding of fas-ligand–containing microvesicles, potentially leading to an evasion of tumor immunity.
Until recently, the metastasis of ovarian cancer has been almost exclusively studied as a process involving individual cells. However, multicellular clusters of self-adherent cells, known as spheroids, can be isolated from the ascitic fluid of women with ovarian cancer. Spheroids readily adhere to both extracellular matrix proteins, such as collagen IV, and mesothelial cells in monolayer culture using beta-1 integrins. Once adherent, the cells contained in spheroids disaggregate, allowing them to invade underlying mesothelial cells and create invasive foci.33 These observations are consistent with the hypothesis that ovarian cancer spheroids play an important role in the metastatic potential of ovarian cancer. Recent evidence has shown that a loss of circulating gonadotropins results in a dose-dependent decrease in the expression of vascular endothelial growth factor (VEGF) in the outer, proliferating cells of ovarian cancer spheroids,34 indicating that these cell clusters remain responsive to signals in their microenvironment that may promote metastasis.
The presence of spheroids in ascites may also help to explain the frequent persistence and frequent recurrence of ovarian cancer after treatment. Spheroids express high levels of p27 and P-glycoprotein that contribute, at least in part, to their relative resistance to the cytotoxic effects of paclitaxel when compared with ovarian cancer cells in monolayer culture. Ovarian cancer spheroids have also been shown to be relatively resistant to the cytotoxic effects of radiation.35 These observations are consistent with in vitro studies that demonstrate that the signals generated by adhesion to specific components of the extracellular matrix, such as collagen IV, can modify the sensitivity of ovarian cancers to chemotherapy. However, the mechanisms by which the aggregation of malignant cells promote or enhance cell survival remain unclear. It is also unclear how the aggregation of these malignant cells might promote or enhance the migration, attachment, or invasion of ovarian cancer cells. Insight into these questions is likely to come from genetic models, such as the migration of the border cell cluster in Drosophila. Analyses of border cell migration indicate that specific shifts in epithelial polarity, known as the epithelial-mesenchymal transition (EMT), and changes in the patterns of signals arising at junctional complexes are necessary for the invasion and migration of epithelial clusters.36 Signals arising from these junctional proteins appear to be integrated by a specific steroid receptor coactivator, known as AIB1 (Amplified in Breast Cancer 1; SRC3). Ironically, overexpression of AIB1 is a frequent feature of ovarian cancers, suggesting that the pathways regulated by this transcriptional coactivator may also play a critical role in promoting ovarian cancer metastasis. Other proteins first identified in Drosophila, such as Snail, also appear to play an important role in regulating the EMT of transformed ovarian epithelia, further lending credence to the utility of this genetic model.
Angiogenesis
Growth of both primary ovarian cancers and their metastases requires the formation of new blood vessels to support adequate perfusion. This process, known as angiogenesis, mechanistically involves both the branching of new capillaries as well as the remodeling of larger vessels. Other processes, such as vasculogenic mimicry, have also been implicated in tumor angiogenesis.
Angiogenesis is tightly regulated by a balance of pro- and antiangiogenic factors. These include growth factors, such as TGF-β, VEGF, and platelet-derived growth factor; prostaglandins, such as prostaglandin E2; cytokines, such as interleukin 8; and other factors, such as the angiopoietins (Ang-1, Ang-2), and hypoxia-inducible factor-1α (HIF-1α). Many of these angiogenic factors have been implicated in ovarian cancer. For example, VEGF is a family of secreted polypeptides with critical roles in both normal development and human disease. Many cancers, including ovarian carcinomas, release VEGF in response to the hypoxic or acidic conditions typical in solid tumors. Near universal, albeit variable, levels of VEGF expression have been reported in ovarian cancers, in which higher levels correlate with advanced disease and poor clinical prognosis.37 Circulating levels of VEGF have also been reported to be higher in the serum of women with ovarian cancers when compared with those with benign tumors. Expression of HIF-1 correlates well with microvessel density in ovarian cancers and has been proposed to up-regulate VEGF expression.38 Culturing ovarian cancer cell lines under hypoxic conditions stimulates the expression of both HIF-1α and VEGF expression in ovarian cancer cell lines; addition of prostaglandin E2 potentiates the ability of hypoxia to induce the expression of both proangiogenic factors.39
Ironically, many of the molecules implicated in regulating angiogenesis in cancer, such as c-met, also regulate other processes critical for cancer metastasis, such as cell migration and invasiveness. Inhibition of PI3K decreases transcription of VEGF in ovarian cancer cells, an effect that is reversed by the forced expression of AKT. Such observations are consistent with reports that hypoxia not only induces angiogenesis, but also increases the invasiveness of ovarian cancer cells.40 Likewise, an acidic environment induces increased interleukin-8 expression in ovarian cancer in a manner dependent on transcription factors AP-1 and nuclear factor-kB–like factor, suggesting that feedback between these pathways may also determine how tumors interact with their external environment. Undoubtedly, better insight into these interactions will help to define the suitability of these molecules as therapeutic targets.
Epigenetics
It has become increasingly apparent that epigenetic events can lead to cancer as frequently as loss of gene function due to mutations or loss of heterozygosity. The overall level of genomic methylation is reduced in cancer (global hypomethylation), but hypermethylation of promoter regions of specific genes is a common event that is often associated with transcriptional inactivation of specific genes.41 This is critical because the silenced genes are often tumor suppressor genes, and their loss of function can be evident in early stages of cancer but can also drive neoplastic progression and metastasis. Epigenetic gene silencing is a complex series of events that includes DNA hypermethylation of CpG islands within gene-promoter regions, histone deacetylation, methylation or phosphorylation, or histone demethylation. Global hypermethylation of CpG islands appears to be prevalent but highly variable in ovarian cancer tissue.42 Multiple genes are abnormally methylated in ovarian cancer compared with normal ovarian tissue, including p16, RAR-β, H-cadherin, GSTP1, MGMT, RASSF1A, leukotriene B4 receptor, MTHFR, progesterone receptor, CDH1, IGSF4, BRCA1, TMS1, estrogen receptor-α, the putative tumor suppressor km23 (TGFB component), and others.43 The degree of DNA methylation and the demethylation activity of chemotherapeutic drugs and the sensitive relations of histone acetylation and the specificity of demethylation of select genes are important to ensure the success of treatment and prevent disease recurrence.
Role of Specific Immune Responses
The novel observation by William Coley in the 1890s that severe bacterial infections could induce an antitumor response in patients with partially resected tumors has evolved into an understanding that the immune system can recognize tumor-associated antigens and direct a targeted response. The concept of “cancer immunoediting” suggests that the immune system not only protects the host against the development of primary cancers but also dynamically sculpts tumor immunogenicity.44 In epithelial ovarian cancer, support for the role of immune surveillance of tumors comes from recent observations that the presence of infiltrating T lymphocytes (TILS) in tumors is associated with improved survival of patients with the disease.45,46 In one study, there was improved survival of patients with higher frequencies of intraepithelial CD8+ TIL (55 months vs. 26 months; hazard ratio [HR] 0.33; 95% CI, 0.18 to 0.60; P = .0003).46 In addition, the subgroups with a high versus low intraepithelial CD8+/CD4+ TIL ratio had a median survival of 74 versus 25 months (HR 0.30; 95% CI, 0.16 to 0.55; P = .0001). This unfavorable effect of CD4+ T cells on prognosis was found to be due to CD25+ Forkhead box P3+ regulatory cells (Treg, suppressor T cells), as indicated by survival in patients with high versus low CD8+/Treg ratios (median, 58 months vs. 23 months; HR 0.31; 95% CI, 0.17 to 0.58; P = .0002).46
Finally, advanced-stage ovarian cancer patients can have detectable tumor-specific cytotoxic T cell and antibody immunity. This was illustrated in a recent study that indicated that immunity to p53 predicted improved overall survival in patients with advanced-stage disease.47 All of these observations support clinical trials of immunotherapy for epithelial ovarian cancer in an effort to elicit effective antitumor responses. Major obstacles include the identification of tumor-restricted immunogenic targets, generation of a sufficient immune response to cause tumor rejection, and approaches to overcome tumor evasion of immune attack.
Ovarian Cancer-Specific Antigens
The development of approaches for analyzing humoral and cellular immune reactivity to cancer in the context of the autologous host has led to the molecular characterization of tumor antigens recognized by autologous CD8+ T cells or antibodies. As a consequence of these advances, human tumor antigens defined to date can be classified into one or more of the following categories: (1) differentiation antigens, such as tyrosinase, Melan-A/MART-1, and gp 100; (2) mutational antigens, such as CDK4, β-catenin, caspase-8, and P53; (3) amplification antigens, such as Her2/neu and p53; (4) splice variant antigens, such as NY-CO-37/PDZ-45 and ING1; (5) viral antigens, such as human papillomavirus (HPV) and Epstein-Barr virus; and (6) cancer-testis antigens, such as MAGE, NY-ESO-1, and LAGE-1. Thus, it is clear that some antigens may play a crucial role in progression of tumor cells (e.g., Her2/neu) and could be useful as biomarkers of disease progression and targets of therapy. On the other hand, in considering an antigenic target for ovarian cancer immunotherapy, an ideal candidate antigen should not only demonstrate high-frequency expression in the tumor tissues and restricted expression in normal tissues, but also provide evidence for inherent immunogenicity. In this regard, the cancer-testis antigens are a distinct and unique class of differentiation antigens with high levels of expression in adult male germ cells, but generally not in other normal adult tissues, and aberrant expression in a variable proportion of a wide range of different cancer types. Among cancer-testis antigens, NY-ESO-1, initially defined by serologic analysis of recombinant cDNA expression (SEREX) libraries in esophageal cancer, is particularly immunogenic, eliciting both cellular and humoral immune responses in a high proportion of patients with advanced NY-ESO-1 expressing ovarian cancer.48
The reasons for the aberrant expression of cancer-testis antigens in cancer are currently unknown. Nevertheless, the fact that the expression of these antigens is restricted to cancers, gametes, and trophoblast suggests a link between cancer and gametogenesis. Although possible mechanisms include global demethylation and histone deacetylation, the induction of a “gametogenic” program in cancer has also been proposed.49 Although several lines of evidence have shown that spontaneous or vaccine-induced tumor-antigen–specific T cells can recognize ovarian cancer targets, prolongation of survival in patients treated with immunization has only rarely been observed. This is probably a reflection of several in vivo immunosuppressive mechanisms in tumor-bearing hosts. A recently described mechanism in ovarian cancer is the expression of inhibitory molecules such as programmed death-1 (PD-1) and lymphocyte activation gene-3 (LAG-3).50 Together, these molecules render ovarian tumor infiltrating CD8+ T cells “hyporesponsive,” wherein effector function is most impaired in antigen-specific LAG-3+PD-1+CD8+ TILs.
ENDOMETRIAL CANCER
The current concept of endometrial cancer integrates histopathology with molecular genetic mechanisms of cancer development. Two major pathogenetic variants of endometrial carcinoma, type I (endometrioid) and type II (serous), evolve via divergent pathways and different precursor lesions, different genetic abnormalities, and ultimately different clinical outcomes parallel their distinct histology.
Type I Cancers
More than 90% of uterine cancers arise in the self-renewing glandular epithelium that lines the uterine cavity. The endometrium epithelium responds to steroid hormones with well-characterized patterns of growth and maturation critical for its role in normal reproduction. Estrogen is a well-recognized growth factor for the endometrium, promoting glandular proliferation. Subsequent exposure to the progestin-rich environment that follows ovulation results in an arrest of endometrial proliferation accompanied by glandular luteinization. Several decades of epidemiologic evidence has convincingly demonstrated that continued, unopposed exposure to estrogen is associated with an increased risk of developing endometrial cancer. These risks are particularly notable among postmenopausal women treated with estrogen-only hormone replacement. Following the introduction of hormone replacement therapy, the incidence of endometrial cancer among women in the United States rose steadily. An association between the growth-promoting effects of estrogen and endometrial carcinomas is thought to underlie the epidemiologic associations found for endometrial cancers, medical conditions such as anovulation, obesity, and other epidemiologically defined risk factors, including early age at menarche and nulliparity.
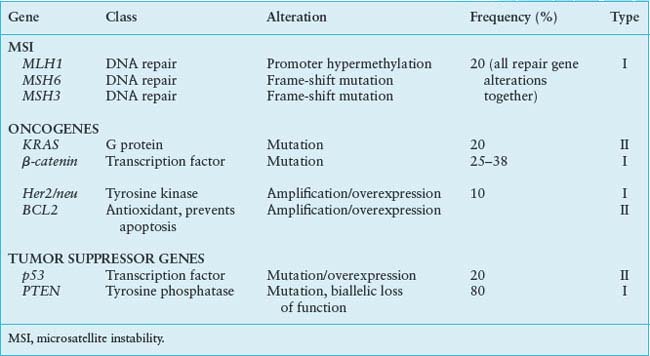
The estrogen-related endometrioid adenocarcinomas account for 80% of endometrial cancer, demonstrate large number of genetic changes, and appear to arise via a progression pathway. Common genetic changes in this type of endometrial carcinoma include microsatellite instability (MSI), or specific mutations of PTEN, K-ras, and βB-catenin genes.
Microsatellite Instability
Microsatellites are short segments of repetitive DNA found predominately in noncoding DNA and scattered through the genome. The MSI phenotype is expressed in the cells with changes in the number of repeat elements as compared with normal tissue because of DNA repair error during replication. Approximately 20% of type I endometrial cancers demonstrate MSI phenotype, while MSI in type II cancers is very rare, present in less than 5% of the cases.51 MSI is due to inactivation of any of the mismatch repair genes and proteins: MLH1, MSH2, MSH3, and MSH6. The most common mechanism of MSI in the endometrium is inactivation of MLH1 by epigenetic silencing of its promoter through hypermethylation of CpG islands, followed by MSH6 mutation and MSH3 frame shift mutations. In contrast, the MSI present in colon cancer is predominantly due to mutations in MSH2, followed by MLH1 and MSH6 mutations. MSI is an early event in type I cancers and it has been described in precancerous lesions. Once established, MSI may specifically target or inactivate genes with susceptible repeat elements, such as TGF-β1 receptors and IGFIIR, resulting in new subclones with altered capacity to invade and metastasize.
PTEN
Inactivation of PTEN (phosphatase and tensin homolog) tumor suppressor gene located at 10q23 is the most common genetic defect in type I endometrial cancers, and it is present in more than 80% of tumors that are preceded by histologically distinct premalignant phase.52 The predominant PTEN activity is a lipid phosphatase that converts inositol triphosphates into inositol biphosphate, thus inhibiting survival and proliferative pathways that are activated by inositol triphosphatase. PTEN protein functions in maintaining G1 arrest and enabling apoptosis via an AKT-dependent mechanism. PTEN inactivation is caused by various mechanisms. The most common PTEN defect in endometrial cancer is its complete loss of function through inactivation of both alleles. Mutations or deletions that result in loss of heterozygosity at PTEN locus are also observed with high frequency. The PTEN mutations pattern is different in microsatellite stable and MSI cancers. MSI tumors have a higher frequency of deletions, involving three or more base pairs, as compared with the microsatellite-stable tumors. In addition, the mutations in MSI tumors only rarely involve the polyadenine repeat of exon 8, which is the expected target.
KRAS
KRAS mutations have been found in up to 30% of type I endometrial cancers. The frequency of KRAS mutations is particularly high in MSI-positive tumors.53
β-catenin
β-catenin (3p21) is a component of the E-cadherin–catenin complex essential for cell differentiation and maintenance of normal tissue architecture, and it also plays a role in signal transduction. The APC protein down-regulates β-catenin levels, inducing phosphorylation of serine-threonine residues coded in exon 3 of the β-catenin and its degradation via ubiquitin-proteosome pathway. Gain of function mutations in β-catenin exon 3 are seen in 25% to 38% of type I cancers.54 These mutations result in protein stabilization, accumulation, and transcriptional activation. β-catenin mutations have been found also in premalignant endometrial lesions. β-catenin changes may characterize pathways of endometrial cancer separate from PTEN mutations and are characterized by squamous differentiation. Several genes may be targets of dysregulated β-catenin pathway. Although in colon cancer elevated β-catenin levels trigger cyclin D1 expression and uncontrolled progression of tumor cells into the cell cycle, in type I endometrial cancers, β-catenin may regulate expression of MMP-7, which has a role in the establishment of microenvironment necessary for maintenance of tumor growth.
Type II Endometrial Cancer
The more aggressive, non–estrogen-related, nonendometrioid cancers (predominantly serous and clear cell carcinomas) are characterized by p53 mutations and Her2/neu amplification and bcl-2 changes. These high-grade tumors are known to be associated in some cases with an identifiable intraepithelial neoplasia component. The same pattern of genetic changes is seen in the preneoplastic atrophic endometrium, suggesting that these are early events in type II tumors carcinogenesis55 (Table 23.2).
CERVIX, VAGINAL, AND VULVAR CANCERS
Role of Human Papillomavirus
Persistent infections with specific high-risk HPV genotypes (e.g., HPV-16, HPV-18, HPV-31, HPV-33, and HPV-45) have been identified as an essential, although not sufficient, factor in the pathogenesis of majority of cancers of the cervix, vagina, and vulva.56 The existence of papilloma viruses was first demonstrated by Shope in the 1930s using an ultrafiltrate of warts from rabbits.57 Since then, papilloma viruses with an epithelial tropism have been demonstrated in nearly every mammalian species, including humans. The HPVs are encapsulated DNA viruses containing a double-stranded DNA genome of approximately 7,800 base pairs. After infecting a suitable epithelium, viral DNA replication takes place in the basal cells of the epidermis, where the HPV genome is stably retained in multiple copies, guaranteeing its persistence in the epithelium’s proliferative cells. This occurs early in preneoplastic lesions, when the viral genome still persists in an episomal state. In most invasive cancers and also in a few high-grade dysplastic lesions, however, integration of high-risk HPV genomes into the host genome is observed. Integration seems to be a direct consequence of chromosomal instability and an important molecular event in the progression of preneoplastic lesions. In a review of more than 190 reported integration loci, HPV integration sites are found to be randomly distributed over the whole genome with a clear predilection for genomic fragile sites. No evidence for targeted disruption or functional alteration of critical cellular genes by the integrated viral sequences could be found.58
TABLE 23.2
GENETIC ALTERATIONS IN ENDOMETRIAL CANCER
The ability of high-risk HPVs to transform human epithelia relates to the transcription of specific viral gene products. Transcription from the HPV genome occurs in two waves: an early phase with seven to eight gene products and a late phase with two gene products (L1, L2). Early-phase gene products play a critical role in viral DNA replication (E1, E8) and regulation of transcription (E2, E8). In contrast, the L1 and L2 genes code for the capsid’s primary and secondary proteins, respectively. The ability of different high-risk HPVs to transform human epithelia has been primarily associated with the expression of two specific viral gene products, E6 and E7. Transformation of human genital tract epithelium likely requires the expression of both E6 and E7; transfection of human keratinocytes in vitro with either is insufficient to accomplish this phenomenon.
At a molecular level, E6 and E7 interfere with important control mechanisms of the cell cycle, apoptosis, and maintenance of chromosomal stability by directly interacting with p53 and pRB, respectively. Moreover, recent studies demonstrated that the two viral oncoproteins cooperatively disturb the mechanisms of chromosome duplication and segregation during mitosis and thereby induce severe chromosomal instability associated with centrosome aberrations, anaphase bridges, chromosome lagging, and breaking.59 They have also been shown to interact with a number of other cellular proteins whose role in epithelial transformation remains unclear, including transcriptional coactivators, such as p300, and components of junctional complexes, such as hDlg1. Altered expression of hDlg1 has been observed in high-grade cervical dysplasias, consistent with the hypothesis that these gene products play an early role in the HPV-induced progression to cervical cancer. Specific sequence differences have been associated with different levels of risk for ultimately developing cervical cancers. For example, recent evidence demonstrates that the sequence of E6 found in Ashkenazi populations confers a protective advantage against developing cervical cancer, previously attributed to the practice of circumcision. Although much less understood, other early genes, such as E2, have also been implicated in the transformation.
Immune Evasion by Human Papillomavirus
HPV infection has a transitory pattern, whereby most individuals (70% to 90%) eliminate the virus 12 to 24 months after initial diagnosis.60 HPV has evolved several strategies to evade immune attack. Most obviously, papillomaviruses do not infect and replicate in antigen-presenting cells that are located in the epithelium, nor do they lyse keratinocytes, so there is no opportunity for antigen-presenting cells to engulf virions and present virion-derived antigens to the immune system. Furthermore, there is no blood-borne phase of infection, so the immune system outside the epithelium has little opportunity to detect the virus. Additionally, HPVs have exploited the redundancy of the genetic code to keep the levels of “late” proteins low.61 Papillomavirus capsid protein production in mammalian cells is markedly up-regulated if the “viral” codons are replaced by the ones that are used by mammals, thereby limiting opportunities for the host to mount an effective immune attack. Following viral integration and subsequent malignant change, the local tumor environment at the cervical lesion is immunosuppressive. Thus, antigen-loaded dendritic cells (DCs) fail to mature, and immature DCs transmit a tolerogenic, rather than an immunogenic, signal to T cells bearing antigen-directed T-cell receptors in draining lymph nodes.
Human Papillomavirus Vaccines
The aim of prophylactic vaccination is to generate neutralizing antibodies against the HPV L1 and L2 capsid proteins. Prophylactic vaccine development against HPV has focused on the ability of the L1 and L2 virion structural proteins to assemble into viruslike particles (VLPs). VLPs mimic the natural structure of the virion and generate a potent immune response. Because the VLPs are devoid of DNA, they are not infectious or harmful. HPV VLPs can be generated by expressing the HPV capsid protein L1 in baculovirus or yeast. They consist of five L1 subunits that multimerize into immunogenic pentamers. Seventy-one L1 pentamers, in turn, multimerize into an HPV VLP. Initial studies have shown that VLPs are capable of inducing high titers of neutralizing antibodies to L1 and L2 epitopes.62 Furthermore, VLPs have proven effective in generating HPV type-specific protection from viral challenge in animal papillomavirus models.
With the approval of preventive HPV vaccines that encompass HPV-16, -18, -6, and -11, large prevention clinical trials targeting the most prevalent HPV types in different regions of the world are warranted. Questions such as the necessity of repeat vaccinations and longevity of protection from HPV infection remain to be determined. It is estimated that if women were vaccinated against all high-risk types of HPV before they become sexually active, there should be a reduction of at least 85% in the risk of cervical cancer, and a decline of 44% to 70% in the frequency of abnormal Papanicolaou (Pap) smears attributable to HPV.63 Unfortunately, even after vaccination is implemented, a reduction in the incidence of cervical cancer could not be expected to become apparent for at least a decade.64 Therefore, therapeutic vaccines are still very much needed to reduce the morbidity and mortality associated with cervical cancer.
The therapeutic approach to patients with preinvasive and invasive cervical cancers is to develop vaccine strategies that induce specific CD8+ cytotoxic T lymphocyte (CTL) responses aimed at eliminating virus-infected or transformed cells. The majority of cervical cancers express the HPV-16-derived E6 and E7 oncoproteins, which are thus attractive targets for T-cell–mediated immunotherapy. Several HPV vaccine strategies have successfully elicited immune responses against HPV E6 and E7 epitopes and have prevented tumor growth on challenge with HPV-16-positive tumor cells in mice. Early-phase human trials using therapeutic vaccines have shown that they are safe, as no serious adverse effects have been reported. Other approaches currently undergoing preclinical development include the use of recombinant alpha viruses such as Venezuelan equine encephalitis virus, Semliki Forest virus, and naked DNA vaccination.
GESTATIONAL TROPHOBLASTIC DISEASE
Gestational trophoblastic disease (GTD) encompasses a diverse group of diseases with unique cytogenetic and molecular pathogenesis. The current concept implies that the abnormal trophoblastic tissue in GTD recapitulates the trophoblast present in the early developing placenta and the implantation site. Complete mole is diploid, usually 46, XX is androgenetic in origin, resulting from duplication of a haploid paternal genome (23X). More than 90% of complete moles contain this DNA content, and the remaining group are also androgenetic except 46, XY, and formed by dispermy. Partial moles are triploid, and diandric, usually XXY (58%), XXX (40%), and XYY (2%). Predominance of paternal chromosomes is therefore characteristics of molar pregnancy. Synergistic up-regulation of CMYC, ERBB2, CFMS, and BCL2 has been suggested in pathogenesis of complete mole,65 and similar findings have been confirmed in choriocarcinoma, while mutational analysis of p53 and KRAS failed to show mutations in either complete mole or choriocarcinoma. The other genes involved in the development of choriocarcinoma include DOC-2, a candidate tumor-suppressor gene and a putative tumor suppressor at 7p12-7q11.23,66 and the RAS guanosine triphosphate hydrolase (GTPase) activating protein. It has been postulated that GTD develops by a mechanism of monoallelic contribution, when the gene susceptible to inactivation would be affected by one-hit kinetics. Alternatively, uniparental transmission of genes that are parentally imprinted would impair their regulation.
Placental-site trophoblastic tumor (PSTT) represents neoplastic transformation of implantation site intermediate trophoblast. Placental-site trophoblastic tumor is characterized by aberrant expression of cyclins and p53. Further efforts are needed for better understanding of persistent trophoblastic disease.
Selected References
The full list of references for this chapter appears in the online version.
1. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004;21(2):137.
3. Levanon K, Crum C, Drapkin R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J Clin Oncol 2008;26:5284.
5. Wright JW, Pejovic T, Fanton J, Stouffer RL. Induction of proliferation in the primate ovarian surface epithelium in vivo. Hum Reprod 2008;23:129.
6. Wright JW, Pejovic T, Lawson M, et al. Ovulation in the absence of the ovarian surface epithelium in the primate. Biol Reprod 2010;82:599.
8. Ganesan S, Richardson AL, Wang ZC, et al. Abnormalities of the inactive X chromosome are a common feature of BRCA1 mutant and sporadic basal-like breast cancer. Cold Spring Harb Symp Quant Biol 2005;70:93.
9. Garcia-Higuera I, Taniguchi T, Ganesan S, et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 2001;7(2):249.
10. Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008;451:1116.
11. Boudsocq F, Benaim P, Canitrot Y, et al. Modulation of cellular response to cisplatin by a novel inhibitor of DNA polymerase beta. Mol Pharmacol 2005;67(5):1485.
12. Kennedy RD, Chen CC, Stuckert P, et al. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest 2007;117:1440.
13. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434:913.
14. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917.
15. Song H, Ramus SJ, Tyrer J, et al. A genome-wide association study identifies a new ovarian cancer susceptibility locus on 9p22.2. Nat Genet 2009;41:996.
17. Meinhold-Heerlein I, Bauerschlag D, Hilpert F, et al. Molecular and prognostic distinction between serous ovarian carcinomas of varying grade and malignant potential. Oncogene 2005;24:1053.
24. Landen CN Jr, Birrer MJ, Sood AK. Early events in the pathogenesis of epithelial ovarian cancer. J Clin Oncol 2008;26:995.
25. Berchuck A, Iversen ES, Lancaster JM, et al. Prediction of optimal versus suboptimal cytoreduction of advanced-stage serous ovarian cancer with the use of microarrays. Am J Obstet Gynecol 2004;190:910.
26. Hu W, Wu W, Nash MA, et al. Anomalies of the TGF-beta postreceptor signaling pathway in ovarian cancer cell lines. Anticancer Res 2000;20(2A):729.
28. van Haaften-Day C, Shen Y, Xu F, et al. OVX1, macrophage-colony stimulating factor, and CA-125-II as tumor markers for epithelial ovarian carcinoma: a critical appraisal. Cancer 2001;92(11):2837.
30. Ren J, Xiao YJ, Singh LS, et al. Lysophosphatidic acid is constitutively produced by human peritoneal mesothelial cells and enhances adhesion, migration, and invasion of ovarian cancer cells. Cancer Res 2006;66(6):3006.
31. Sengupta S, Kim KS, Berk MP, et al. Lysophosphatidic acid down-regulates tissue inhibitor of metalloproteinases, which are negatively involved in lysophosphatidic acid-induced cell invasion. Oncogene 2007;26:2894.
32. Do TV, Symowicz JC, Berman DM, et al. Lysophosphatidic acid down-regulates stress fibers and up-regulates pro-matrix metalloproteinase-2 activation in ovarian cancer cells. Mol Cancer Res 2007;5(2):121.
33. Burleson KM, Hansen LK, Skubitz AP. Ovarian carcinoma spheroids disaggregate on type I collagen and invade live human mesothelial cell monolayers. Clin Exp Metastasis 2004;21(8):685.
34. Schiffenbauer YS, Abramovitch R, Meir G, et al. Loss of ovarian function promotes angiogenesis in human ovarian carcinoma. Proc Natl Acad Sci U S A 1997;94(24):13203.
36. Szafranski P, Goode S. A Fasciclin 2 morphogenetic switch organizes epithelial cell cluster polarity and motility. Development 2004;131(9):2023.
37. Kassim SK, El-Salahy EM, Fayed ST, et al. Vascular endothelial growth factor and interleukin-8 are associated with poor prognosis in epithelial ovarian cancer patients. Clin Biochem 2004;37(5):363.
41. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer—a mechanism for early oncogenic pathway addiction? Nat Rev Cancer 2006;6(2):107.
42. Wei SH, Chen CM, Strathdee G, et al. Methylation microarray analysis of late-stage ovarian carcinomas distinguishes progression-free survival in patients and identifies candidate epigenetic markers. Clin Cancer Res 2002;8(7):2246.
44. Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol 2006;90:1.
45. Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 2003;348(3):203.
46. Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 2005;102(51):18538.
47. Goodell V, Salazar LG, Urban N, et al. Antibody immunity to the p53 oncogenic protein is a prognostic indicator in ovarian cancer. J Clin Oncol 2006;24(5):762.
48. Odunsi K, Jungbluth AA, Stockert E, et al. NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer Res 2003;63(18):6076.
49. Old LJ. Cancer/testis (CT) antigens—a new link between gametogenesis and cancer. Cancer Immunity 2001;1:1.
50. Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A 2010;107:7875.
51. Mutter GL, Boynton KA, Faquin WC, Ruiz RE, Jovanovic AS. Allelotype mapping of unstable microsatellites establishes direct lineage continuity between endometrial precancers and cancer. Cancer Res 1996;56(19):4483.
52. Mutter GL, Lin MC, Fitzgerald JT, et al. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst 2000;92(11):924.
54. Mirabelli-Primdahl L, Gryfe R, Kim H, et al. Beta-catenin mutations are specific for colorectal carcinomas with microsatellite instability but occur in endometrial carcinomas irrespective of mutator pathway. Cancer Res 1999;59(14):3346.
56. zur Hausen H. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J Natl Cancer Inst 2000;92(9):690.
58. Wentzensen N, Vinokurova S, von Knebel Doeberitz M. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res 2004;64(11):3878.
60. Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med 1998;338(7):423.
62. Koutsky LA, Ault KA, Wheeler CM, et al. A controlled trial of a human papillomavirus type 16 vaccine. N Engl J Med 2002;347(21):1645.
63. Walboomers JM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999;189(1):12.
66. Matsuda T, Sasaki M, Kato H, et al. Human chromosome 7 carries a putative tumor suppressor gene(s) involved in choriocarcinoma. Oncogene 1997;15(23):2773.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


