INTRODUCTION
SUMMARY
The understanding of hematology is dependent upon an appreciation of genetic principles and the tools that can be used to study genetic variation.* All the genetic information that makes up an organism is encoded in the DNA. This information is transcribed into mRNA, and then the triplet code of those mRNAs is translated into protein. Changes that affect the DNA or RNA sequence or its expression, either in the germline or acquired after birth, can cause hematologic disorders. These may be mutations that change the DNA sequence, including single base changes, deletions, insertions, and duplications, or they may be epigenetic changes that affect gene expression without any change in the DNA sequence.
The detection of mutations that cause a variety of diseases is now possible and has become a routine method for the diagnosis of some disorders. Large-scale DNA sequencing can be used to identify disease-causing genes and to carry out genetic testing. The development of methods to disrupt or prevent expression of specific genes has made it possible to produce mouse models of human hematologic diseases, and such models have the potential to serve as means to better understand pathophysiology and to study treatment strategies.
Inheritance patterns depend upon the biologic effect and chromosomal location of the mutation. Common autosomal recessive hematologic diseases include sickle cell disease, the thalassemias, and Gaucher disease. Hereditary spherocytosis, thrombophilia caused by factor V Leiden, most forms of von Willebrand disease, and acute intermittent porphyria are characterized by autosomal dominant inheritance. Mutations that cause glucose-6-phosphate dehydrogenase deficiency, hemophilias A and B, and the most common form of chronic granulomatous disease, are all carried on the X chromosome and, therefore, manifest X-linked inheritance, with transmission of the disease state from a heterozygous mother to her son. Understanding the genetics of a disorder is necessary for accurate genetic counseling.
Acronyms and Abbreviations:
BACs, bacterial artificial chromosomes; bp, base pairs; cDNA, complementary DNA; CNV, copy number variant; CpG, cytosine phosphate guanine; ENU, N-ethyl-N-nitrosourea; G6PD, glucose-6-phosphate dehydrogenase; HNPCC, hereditary nonpolyposis colorectal cancer; lncRNA, long noncoding RNA; miRNA, microribonucleic acid; mRNA, messenger ribonucleic acid; mtDNA, mitochondrial DNA; NADH, nicotinamide adenine dinucleotide (reduced form); PACs, P1-derived artificial chromosomes; PCR, polymerase chain reaction; RISC, RNA-induced silencing complex; RNAi, RNA interference; rRNA, ribosomal ribonucleic acid; RT-PCR, reverse transcriptase polymerase chain reaction; SCID, severe combined immunodeficiency; siRNA, small interfering ribonucleic acid; SNP, single nucleotide polymorphism; STR, short tandem repeat; tRNA, transfer ribonucleic acid; YAC, yeast artificial chromosome.
*In the previous edition, this chapter was written by Ernest Beutler and portions of that chapter have been retained.
GENETICS AND HEMATOLOGIC DISORDERS
Many of the hematologic diseases described in this text have a genetic basis. Often the disease is caused by a mutation in a single gene. Some of these disorders, such as sickle cell disease (Chap. 49), thalassemia (Chap. 48), glucose-6-phosphate dehydrogenase (G6PD) deficiency (Chap. 47), and factor V Leiden (Chap. 130), are common, whereas others, such as congenital dyserythropoietic anemia type I (Chap. 39), chronic granulomatous disease (Chap. 66), and afibrinogenemia (Chap. 125), are rare. All are caused by mutations in a gene that result in the formation of a defective protein or an insufficient amount of a normal protein. The principal focus of this chapter is such genetic disorders. However, a number of acquired hematologic diseases, including lymphomas, leukemias, and other clonal hematologic diseases, are the consequence of acquired damage to the genetic apparatus. Understanding these diseases requires an appreciation of how the genetic apparatus functions.
All of the information required for the development of a complete adult organism is encoded in the DNA of a single cell—the zygote. This information, designated the genome, includes the data needed for the synthesis of all enzymes; all the plasma proteins, including the clotting factors, complement components, and the transport proteins; all the membrane proteins, including receptors; and all of the cytoskeletal proteins. The units of information into which the genome is organized are the genes, which are composed of sequences of DNA. By serving as the blueprints of proteins in the body, genes ultimately influence all aspects of body structure and function. There are approximately 21,000 protein-coding genes and an additional 10,000 genes that do not encode proteins but affect the regulation of genes.1 An error in one of these genes often leads to a recognizable genetic disease. To date, more than 20,000 genetic traits and diseases have been identified and cataloged.
DNA has three basic components: the pentose sugar molecule, deoxyribose; a phosphate molecule; and four types of nitrogenous bases. Two of the bases, cytosine and thymine, are single carbon-nitrogen rings called pyrimidines. The other two bases, adenine and guanine, are double carbon-nitrogen rings called purines. The four bases are commonly represented by their first letters: A, C, T, and G.
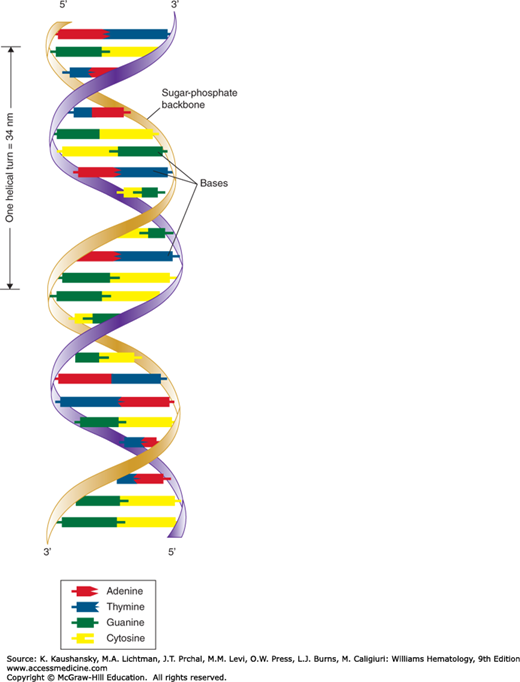
Watson and Crick demonstrated how these molecules are physically assembled together as DNA, proposing the double–helix model, in which DNA appears like a twisted ladder with chemical bonds as its rungs (Fig. 10–1).2,3 The two sides of the ladder are made up of the sugar and phosphate molecules, held together by strong phosphodiester bonds. Projecting from each side of the ladder, at regular intervals, are the nitrogenous bases. The base projecting from one side is bound to the base projecting from the other by a weak hydrogen bond. Therefore, the nitrogenous bases form the rungs of the ladder; adenine pairs with thymine, and guanine pairs with cytosine. Each DNA subunit—consisting of one deoxyribose molecule, one phosphate group, and one base—is called a nucleotide.
Figure 10–1.
Watson-Crick model of the DNA molecule. The DNA structure illustrated here is based on that published by James Watson and Francis Crick in 1953. Note that each side of the DNA molecule consists of alternating sugar and phosphate groups. Each sugar group is united to the sugar group opposite it by a pair of nitrogenous bases (adenine-thymine or cytosine-guanine). The sequence of these pairs constitutes a genetic code that determines the structure and function of a cell. (Reproduced with permission from Patton KT, Thibodeau GA: Anatomy & physiology, 8th edition. St. Louis, MO:Mosby/Elsevier; 2013.)
DNA directs the synthesis of all the body’s proteins. Proteins are composed of one or more polypeptides (intermediate protein compounds), which are, in turn, composed of sequences of amino acids. The body contains 20 different types of amino acids, which are specified by the four nitrogenous bases. To specify (code for) 20 different amino acids with only four bases, different combinations of bases, occurring in groups of three, are used. These triplets of bases are known as codons. Each codon specifies a single amino acid in a corresponding protein. Because there are 64 (4 × 4 × 4) possible codons but only 20 amino acids, there are many cases in which several codons correspond to the same amino acid.
The genetic code is universal: All living organisms use precisely the same DNA codes to specify proteins except for mitochondria, the cytoplasmic organelles in which cellular respiration takes place—they have their own extranuclear DNA. Several codons of mitochondrial DNA encode different amino acids than do the same nuclear DNA codons.
DNA replication consists of breaking the weak hydrogen bonds between the bases, leaving a single strand with each base unpaired. The consistent pairing of adenine with thymine and of guanine with cytosine, known as complementary base pairing, is the key to accurate replication. The unpaired base attracts a free nucleotide only if the nucleotide has the proper complementary base. When replication is complete, a new double-stranded molecule identical to the original is formed. The single strand is said to be a template, or molecule on which a complementary molecule is built, and is the basis for synthesizing the new double strand.
Several different proteins are involved in DNA replication. The most important of these proteins is an enzyme known as DNA polymerase. This enzyme travels along the single DNA strand, adding the correct nucleotides to the free end of the new strand and checking to make sure that its base is actually complementary to the template base. This mechanism of DNA proofreading substantially enhances the accuracy of DNA replication.
A mutation is any inherited alteration of genetic material. Mutations may cause disease or be subtle, silent substitutions that do not change amino acids. A base pair change that alters a single amino acid is termed a missense mutation, and a base pair change that produces a premature stop codon is termed a nonsense mutation. Because they typically result in a complete loss of gene product, nonsense mutations usually produce a more-severe disease phenotype than do missense mutations. For example, nonsense mutations in the factor VIII gene are much more likely to produce severe hemophilia A than are missense mutations. This is an example of a genotype-phenotype correlation, which can be useful in predicting the severity of disease. A frameshift mutation involves the insertion or deletion of one or more base pairs of the DNA molecule. These mutations change the entire “reading frame” of the DNA sequence because the deletion or insertion is not a multiple of three base pairs (bp; the number of base pairs in a codon). Frameshift mutations can thus greatly alter the amino acid sequence and typically lead to a premature stop codon downstream of the mutation. (“In-frame” insertions or deletions, in which a multiple of three bases is inserted or lost, tend to have less-severe disease consequences than do frameshift mutations.) Splice-site mutations describe alterations of the DNA sequence at intron–exon boundaries (see section on“Genes to Proteins” above). These result in a mature mRNA transcript that contains introns or lacks exons. Table 10–1 gives examples of hematologic diseases caused by different types of mutations.
Mutations are rare events. The rate of spontaneous mutations (those occurring in the absence of exposure to known mutagens) in humans is approximately 10−4 to 10−6 per gene per generation. This rate varies from one gene to another, with larger mutation rates for larger genes. At the nucleotide level, the human mutation rate is approximately 10−8 per nucleotide per generation.4,5 Certain DNA sequences have particularly high mutation rates and are known as mutational hot spots. In particular, sequences consisting of a cytosine base followed by a guanine base (CpG) are highly susceptible to mutation and are known to account for a disproportionately large percentage of disease-causing mutations.
DNA is formed and replicated in the cell nucleus, but protein synthesis takes place in the cytoplasm. The DNA code is transported from nucleus to cytoplasm, and subsequent protein is formed through two basic processes: transcription and translation. These processes are mediated by RNA, which is chemically similar to DNA except that the sugar molecule is ribose rather than deoxyribose, and uracil rather than thymine is one of the four bases. The other bases of RNA, as in DNA, are adenine, cytosine, and guanine. Uracil is structurally similar to thymine, so it also can pair with adenine. Whereas DNA usually occurs as a double strand, RNA usually occurs as a single strand.
In transcription, RNA is synthesized from a DNA template, forming messenger RNA (mRNA). RNA polymerase binds to a promoter site, a sequence of DNA that specifies the beginning of a gene. RNA polymerase then separates a portion of the DNA, exposing unattached DNA bases. One DNA strand then provides the template for the sequence of mRNA nucleotides.
The sequence of bases in the mRNA is thus complementary to the template strand, and except for the presence of uracil instead of thymine, the mRNA sequence is identical to the other DNA strand. Transcription continues until a termination sequence is reached. Then the RNA polymerase detaches from the DNA, and the transcribed mRNA is freed to move out of the nucleus and into the cytoplasm.
When the mRNA is first transcribed from the DNA template, it reflects exactly the base sequence of the DNA. In eukaryotes, many RNA sequences are removed by nuclear enzymes, and the remaining sequences are spliced together to form the functional mRNA that migrates to the cytoplasm. The excised sequences are called introns, and the sequences that are left to code for proteins are called exons.
In translation, RNA directs the synthesis of a polypeptide, interacting with transfer RNA (tRNA), a cloverleaf-shaped strand of approximately 80 nucleotides. The tRNA molecule has a site where an amino acid attaches. The three-nucleotide sequence at the opposite side of the cloverleaf is called the anticodon. It undergoes complementary base pairing with an appropriate codon in the mRNA, which specifies the sequence of amino acids through tRNA.
The site of actual protein synthesis is in the ribosome, which consists of roughly equal parts of protein and ribosomal RNA (rRNA). During translation, the ribosome first binds to an initiation site on the mRNA sequence and then binds to its surface, so that base pairing can occur between tRNA and mRNA. The ribosome then moves along the mRNA sequence, processing each codon and translating an amino acid by way of the interaction of mRNA and tRNA.
The ribosome provides an enzyme that catalyzes the formation of covalent peptide bonds between the adjacent amino acids, resulting in a growing polypeptide. When the ribosome arrives at a termination signal on the mRNA sequence, translation and polypeptide formation cease; the mRNA, ribosome, and polypeptide separate from one another; and the polypeptide is released into the cytoplasm to perform its required function.
All cells receive the same complement of genes. Nonetheless, some proteins are tissue-specific. Several circumstances can account for this. Some enzymes that appear to perform the same function are encoded by different genes in different tissues. For example, the pyruvate kinase of leukocytes and that of erythrocytes are under separate genetic control (Chap. 47). In other cases, alternative splicing of the primary mRNA can produce different polypeptides, a phenomenon that is particularly prominent with some of the red cell membrane proteins. Differences in posttranslational processing, including proteolysis and glycosylation of the same polypeptide by different enzymes in different tissues, can lead to different final products. However, in most instances a mutation that affects an enzyme in one type of blood cell will also affect the same enzyme in other blood cells, in liver, in brain, and in other tissues.
Traits caused by single genes are called mendelian traits (after Gregor Mendel). Each gene occupies a position along a chromosome known as a locus. The genes at a particular locus can take different forms (i.e., they can be composed of different nucleotide sequences) called alleles. A locus that has two or more alleles that each occurs with an appreciable frequency (classically defined as 1%) in a population is said to be polymorphic (or a polymorphism). Polymorphisms that involve a single nucleotide are termed single nucleotide polymorphisms (SNPs), while those that involve the presence or absence of larger pieces of DNA are termed copy number variants (CNVs).6,7 Sometimes genetic variants, such as the alleles responsible for sickle cell disease, thalassemia, or G6PD deficiency, reach polymorphic levels because the deleterious effects that they may have are counterbalanced by beneficial effects on survival, such as increased resistance to malaria.8 They are known as balanced polymorphisms. Short tandem repeats (STRs) are a special form of polymorphism consisting of differing numbers of repeating units of one to six nucleotides, for example, ATATATATAT. Such sequences are unstable in evolution of a species and tend to be very polymorphic. Instead of only two possible genotypes, as in the case of most SNPs, there may be 5, 10, or more different numbers of repeats at a given locus in different individuals. As a result, STRs are very useful in genetic mapping and in forensic analysis.9 In addition, an expanded number of repeat copies of some STRs located within or near genes is an important cause of inherited disease.10
Because humans are diploid organisms, each chromosome is represented twice, with one member of the chromosome pair contributed by the father and one by the mother. At a given locus, an individual has one allele whose origin is paternal and one whose origin is maternal. When the two alleles are identical, the individual is homozygous at that locus. When the alleles are not identical, the individual is heterozygous at that locus. The composition of genes at a given locus is known as the genotype. The outward appearance of an individual, which is the result of both genotype and environment, is the phenotype.
In his experiments with garden peas, Gregor Mendel established that many traits can be either dominant or recessive. In dominant traits, such as von Willebrand disease or porphyria cutanea tarda type II, one copy of a disease-causing allele is sufficient for disease causation, so heterozygotes are typically affected. In recessive traits, such as β-thalassemia, two copies of the disease-causing allele must be present, so the affected individual is a homozygote. A carrier is an individual who has a disease gene but is phenotypically normal. Many alleles for a recessive disease occur in heterozygotes that carry one copy of the gene but do not express the disease. When recessive alleles are lethal in the homozygous state, they are eliminated from the population when they occur in homozygotes. By “hiding” in carriers, however, recessive genes for diseases are passed on to the next generation (the word “recessive” comes from the Latin for “hidden”).
Crossing over during meiosis usually occurs with great precision. Homologous genes pair with each other, and although genes that were together on the chromosome before meiosis may now be on opposite chromosomes of the pair, each chromosome still contains a complete set of genes (see Fig. 10–1). Occasionally, however, an error occurs and pairing during meiosis is imperfect. Under these circumstances—unequal crossing over (see Fig. 10–5)—one of the daughter chromosomes contains a duplicated gene, while the other one exists with a gene deleted. Once a duplication has occurred, further duplications occur more readily, because pairing of the first of the duplicate genes on one chromosome with the second gene of the duplicate on the other produces one chromosome with a triplicated gene and one with a single gene (Chap. 48). Duplication has probably played a very important role in the course of evolution11 because the presence of two genes with the same function allows experiments of nature: Mutations can accumulate on one of the genes while the original function is still provided by the duplicate. Examples of the results of gene duplication abound in hematology, particularly with respect to the hemoglobin loci. The γ-chain loci are duplicated, and there are also two nearly identical copies of the α-chain locus (Chap. 48). Furthermore, the close similarity of their amino acid sequence and the fact that they are tightly linked indicate that the β, γ, and δ loci represent the result of duplication of a single ancestral gene. The process of unequal crossing over takes place not only between genes, but also within genes. When this occurs, one would anticipate that a portion of the amino acid sequence of a protein is represented twice on one chromosome and is missing on the other. The Lepore hemoglobins, leading to a thalassemic clinical state, are an example of this type of unequal crossing over (Fig. 48–8). These abnormal hemoglobins have the amino acid sequence of the δ chain at the amino end, and the sequence of the β chain at the carboxyl end. The complement to this kind of abnormality, the “anti-Lepore” hemoglobin, also has also been found (Chap. 49). Similarly, a mutation of the glucocerebrosidase gene causing Gaucher disease has been found to be the result of a crossover between the active gene and the pseudogene.12 The two types of haptoglobin represent an ancestral gene and one in which a major part of that gene has been duplicated.13
TRANSMISSION OF GENETIC DISEASES
The known single-gene diseases can be classified into four major modes of inheritance: autosomal dominant, autosomal recessive, X-linked dominant, and X-linked recessive.14 The first two types involve genes known to occur on the 22 pairs of autosomes. The last two types occur on the X chromosome; very few disease-causing genes are found on the Y chromosome.
The pedigree chart summarizes family relationships and shows which members of a family are affected by a genetic disease (Fig. 10–2).15 Generally, the pedigree begins with one individual in the family, the proband. This individual is usually the first person in the family diagnosed or seen in a clinic.

Diseases caused by autosomal dominant genes are rare, with the most common occurring in fewer than 1 in 500 individuals. Therefore, it is uncommon for two individuals who are both affected by the same autosomal dominant disease to produce offspring together. Affected offspring are usually produced by the union of a normal parent with an affected heterozygous parent. The affected parent can pass either a disease gene or a normal gene to the next generation. On average, half the children will be heterozygous and will express the disease, and half will be normal.
The pedigree in Fig. 10–2
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


