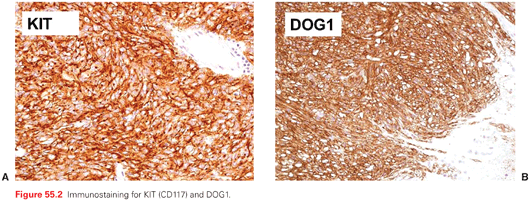
Immunohistochemically, the hallmark of most GISTs is their positivity for KIT (CD117) and DOG-1 (Fig. 55.2 A,B).25–27 A low proportion of GISTs are CD117 negative, which is typical of PDGFRA-mutated GISTs, but immunohistochemical status does not reflect the mutational status with regard to KIT and PDGFRA, per se, so that it has no concrete predictive value for sensitivity to TKIs. Thus, CD117 has only a meaning in the pathologic differential diagnosis. Given their morphology, GISTs must be differentiated from other soft tissue tumors of the gastrointestinal wall, including those of smooth muscle and neural origin and desmoid-type fibromatosis, endocrine tumors, melanocytic tumors, lymphomas, etc. Desmin is rarely positive, as opposed to vimentin and CD34. A negative stain for SDHB identifies the subgroup of SDH-deficient WT GISTs.28,29
Molecularly, GIST have become a relatively heterogeneous and complex group of lesions.30 Gain-of-function mutations of the oncogenes located on chromosome 4 (4q12) coding for the type III receptor tyrosine kinases KIT and PDGFRA can be found in approximately 80% of GISTs.5,6,31 Pathogenetically, they are the drivers of the disease and, therapeutically, underlie the efficacy of currently used TKIs. They are mutually exclusive and result in the constitutive activation of either KIT or PDGFRA, which normally are autoinhibited, being activated by the binding of their respective ligands (i.e., stem-cell factor [Steel factor] and platelet-derived growth factor A). The activation of the receptor binds two molecules of KIT or PDGFRA (dimerization), giving rise to downstream oncogenic signaling, which for both KIT and PDGFRA involves the RAS/MAPK and the PI3K/AKT/mammalian target of rapamycin (mTOR) pathways (Fig. 55.3). Mutations can be deletions, insertions, and missense mutations. They affect: exon 11 of the KIT oncogene, encoding for the juxtamembrane domain of the KIT receptor, in slightly less than 70% of GISTs; exon 9 of KIT, encoding for the extracellular domain of the receptor, in less than 10%; exon 13 and 17 of KIT, encoding for the intracellular ATP-binding pocket and activation loop domains, respectively, in a small minority of GISTs. Approximately 10% of GIST have mutations homologous to these, which affect PDGFRA (i.e., exon 12, 14, and 18 of the oncogene, with 70% being represented by the exon 18 D842V mutation). The latter is known for its wide lack of sensitivity to available TKIs, along with a few other rare exon 18 mutations, whereas the deletion of codons 842 to 845 is sensitive. Possibly because of their similarity with different kinds of normal interstitial cell of Cajal, some tumor cell mutations correlate with elective primary sites of origin. In particular, exon 9 mutations of KIT are preferably found in the small bowel, and PDGFRA mutations are found in the stomach.
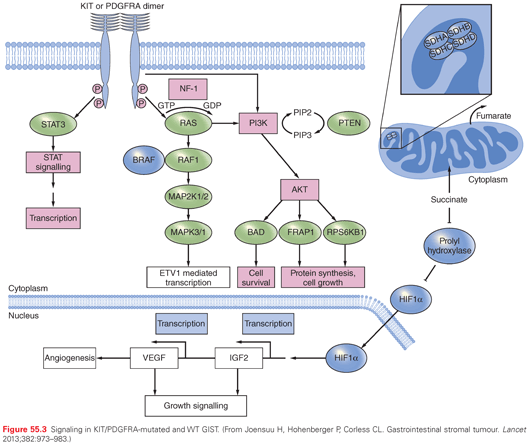
Approximately 10% to 15% of GISTs are WT for KIT and PDGFRA. They make up a family of tumor subsets with different pathogenetic backgrounds and, to some extent, different natural histories (see Fig. 55.3). Their classification is evolving.5,6,31 In essence, as of today, one may identify: (1) SDH-deficient GISTs; (2) neurofibromatosis (NF)-1–related GISTs; or (3) others, including those with the BRAF V600E mutation. In fact, half of WT GISTs are marked by alterations involving the SDH complex, which is crucial for the Krebs cycle and mitochondrial respiratory cell function. Immunohistochemically, these GIST are negative to SDHB staining. A group of them includes pediatric GISTs and can be associated with the Carney triad.13 In fact, these GISTs tend to arise in children and young adults of the female sex, are gastric and multifocal, can metastasize to lymph nodes, have a rather indolent evolution. When the Carney triad is fully expressed, it includes GISTs, pulmonary chondromas, and paragangliomas. Given the absence of mutations to the SDH complex, a posttranscriptional defect leading to dysfunctions of the SDH complex may be in place. These GISTs are SDHA positive. On the other hand, a group of SDH-deficient GISTs carries germ-line mutations of the SDHA, SDHB, or SDHC units of the SDH complex32,33 and may be related to the Carney-Stratakis syndrome.14 This is marked by GIST and paragangliomas. Immunohistochemically, GISTs with SDHA mutations are negative to SDHA staining. The median age of these patients is somewhat higher and the female to male predominance is lower, but the course of disease is indolent as well. Then, WT SDHB-positive GISTs can occur in the context of NF-1, and their pathogenetic mechanism is supposed to be the absence of neurofibromin (i.e., the product of the NF-1 gene), which is mutated.34,35 This may lead to increased activity of the RAS pathway. GISTs related to NF-1 are typically multicentric as well, and have a rather indolent course, but arise from the small bowel. Of course, NF-1 may coexist with a non–NF-1-related GISTs. Finally, the remaining SDHB-positive GISTs are probably a basket of different conditions: some were reported to have the V600E mutation of BRAF36,37 or, more rarely, HRAS, NRAS, and PIK3 mutations.5 All this makes the so-called WT GISTs a variegated family of tumors, which can now be identified not only through a negative definition (i.e., by the lack of KIT and PDGFRA mutations), but through immunohistochemical or cytogenetic markers, pointing to specific subsets with different natural histories.
A very rare subset of familial GISTs does exist, being marked by mutations of KIT or PDGFRA affecting the germ line.16,17 They parallel mutations found in sporadic GISTs and lead to the multicentric and multifocal occurrence of GISTs. The behavior of these GISTs is variable (i.e., it is often indolent but some lesions turn out to become aggressive). Hyperplasia of interstitial cells of Cajal can be found, which may entail altered motility of the gastrointestinal tract. Urticaria pigmentosa and other alterations of skin pigmentation may complete the syndrome.
It is then clear how important genotyping has become for GIST patients. In fact, genotyping has an obvious predictive value, which is crucial for all patients who are candidates for medical therapy, whether in the advanced or in the adjuvant setting. In addition, genotyping has prognostic implications, at least given the peculiar natural history of WT GISTs. Finally, genotyping confirms the pathologic diagnosis in KIT/PDGFRA-mutated GIST, or leads to further pathologic and molecular assessments in WT GISTs. Thus, although there are subsets of GISTs with such a low risk of relapse as not to make them candidates for any medical therapy, a mutational analysis is currently felt as a companion to virtually any pathologic diagnosis of GISTs.
GISTs are rare cancers. Therefore, population-based screening policies are unforeseeable. As for all rare cancers, the clinical aim should be a timely diagnosis in the individual patient with symptoms and/or signs of disease. A difficulty thereof is the anatomical tendency of GIST lesions to grow outwards from the gastrointestinal wall, so that they may go undetected for long periods even when endoscopically explored. However, endoscopic procedures carried out for other reasons may lead to some risk of overdiagnosis, even in such a rare disease, when small gastric lesions are incidentally detected. Some of them will be benign entities, and others will be GISTs unlikely to ever grow as to become clinically relevant. Only a minority of them will turn out to be clinically aggressive GISTs caught in their making.
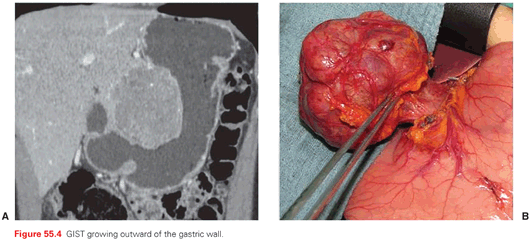
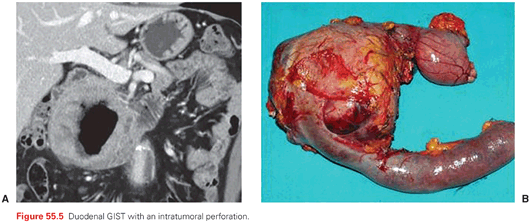
The outward growth of many GISTs (Fig. 55.4) within the gastrointestinal wall is one of the reasons why several are diagnosed relatively late, either as major abdominal masses or as causes of gastrointestinal bleeding, hemoperitoneum, perforations (Fig. 55.5). Therefore, as many as one-fourth of GISTs are diagnosed in a clinical emergency, often leading to surgical explorations resulting in the unexpected finding of the disease. One-fourth of GISTs are discovered incidentally during diagnostic assessments (whether an endoscopic procedure, ultrasound, or computed tomography [CT] scan) done for other reasons. The remaining are diagnosed because of symptoms of compression from an abdominal mass, or chronic anemia, fatigue, and the like. Therefore, GISTs should be included in the differential diagnosis of abdominal masses. When their pertinence to the gastrointestinal wall is clear, the possibility of a GIST may be obvious, with a differential diagnosis mainly against epithelial tumors, small bowel endocrine tumors, lymphomas, paragangliomas, etc. Otherwise, retroperitoneal sarcomas and desmoid-type fibromatosis, germ cell tumors, and lymphomas are the main alternatives. Notably, when this is the clinical presentation, surgery is of choice only for some of the possible alternatives within the clinical differential diagnosis. In addition, preoperative treatments may be resorted to even in some of the surgical indications. On top of this, an intraoperative pathologic differential diagnosis is prohibitive. In principle, therefore, a diagnostic core needle biopsy is suggested by many, allowing pathologic diagnosis and, in the case of GIST, a mutational analysis, prior to any surgical exploration. In the case of gastric or rectal lesions, a biopsy can be carried out by means of endoscopic ultrasound, although, for gastric tumors, the risk of perforation should be factored in depending on the presentation. A CT/ultrasound-guided percutaneous biopsy is the other option, apparently with a negligible risk of dissemination if done at a center of expertise, again factoring in the clinical presentation.38 There may remain some cases in which the difficulty of an endoscopic or percutaneous biopsy and the easiness of a surgical exploration would suggest the latter. In general, however, a biopsy prior to any therapeutic planning can minimize the number of abdominal masses undergoing futile surgery.
Follow-ups after potentially eradicating surgery is aimed at picking up relapses at an early stage. Local relapses are infrequent and tend to develop outwards from the gastrointestinal wall: therefore, an endoscopy is generally not used as a routine follow-up procedure. A CT scan is the most sensitive exam to pick up peritoneal and liver metastases and is recommended. It can be replaced by magnetic resonance imaging (MRI), while ultrasound is much less sensitive on the peritoneum. The maximum risk interval averages 2 to 3 years after surgery or, if an adjuvant therapy was done, after its completion. Long-term relapses are unlikely, although they are occasionally observed, especially in GISTs with a low mitotic rates. All this helps drive rational follow-up policies for potentially cured patients, though there is a lack of any empirical evidence of their effectiveness.39
Conventional stage classification is seldom used.40 Clinicians mainly distinguish localized from metastatic disease and, if the disease is localized and amenable to complete surgery, quantify the risk of relapse.20,22,24
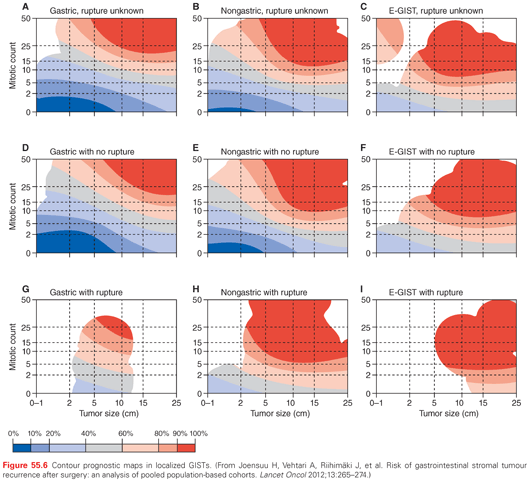
Current risk classification systems are based on the combination of mitotic count, tumor size, and site of origin. Indeed, the mitotic count is the main prognostic factor, proportionally correlating to the risk of relapse. Its downside has turned out to be its possibly low reproducibility rate, but clearly this can be higher if the pathologist is aware of its importance in driving treatment choices. Tumor size is the next prognostic factor. On one side, it singles out very small gastric lesions (<2 cm), which may undergo watchful surveillance if incidentally discovered endoscopically. On the other, it highlights lesions in excess of 5 to 10 cm, which have a worse prognosis. With regard to the primary site, gastric lesions have a better prognosis than small bowel and rectal GISTs. Thus, the combination of these three factors allows one to forecast a risk of relapse by using tools such as the Armed Forces Institute of Pathology (AFIP) risk classification, the Memorial Sloan Kettering Cancer Center (MSKCC) nomogram, or the contour maps. The contour maps have the advantage of treating both the mitotic rate and tumor size as continuous variables as they are, so that the accuracy is increased especially for intermediate-risk cases (Fig. 55.6). Also, reproducibility issues become less crucial by factoring mitotic count as a continuous variable. In addition, contour maps segregate the prognosis of lesions that underwent tumor rupture, which is a highly adverse prognostic factor in diseases anatomically facing the peritoneum.41
The natural history of advanced GISTs is marked by their potential extension to the peritoneum and/or the liver. Thus, a CT scan is the staging procedure of choice to rule out metastatic disease. Lung metastases are rare, with the possible exception of rectal GISTs, although a chest CT scan is generally used to extend a staging workup to lungs and the mediastinum. Bone metastases are possible, but they are usually confined to the very advanced stages of disease, so that the skeleton is not routinely assessed in the lack of symptoms.42 Other sites of distant metastases are exceedingly rare. Lymph node regional metastases are not typical of GISTs, as for mesenchymal tumors in general, with the remarkable exception of WT GISTs occurring in children and/or within syndromes. In addition, all syndromic GISTs may be multifocal and multicentric.43,44 This is not tantamount to metastatic spread, being rather a marker of their inherent natural history. All these features of the natural history of GISTs drive staging procedures, in addition to the potential for other syndromic correlates, depending on the presentation.
Localized GISTs with no evidence of distant metastases are treated with surgery, followed by adjuvant medical therapy if the risk of relapse is significant. This treatment strategy capitalizes on the consolidated curative potential of surgery and prolongs the relapse-free interval of patients who are not eradicated. When surgery is unfeasible or could be made less mutilating or easier through downsizing, medical therapy is used if the genotype is sensitive to imatinib, possibly followed by surgery and the completion of a medical adjuvant treatment if the risk of relapse is significant. When the disease is metastatic, medical therapy with TKIs is standard treatment and should be maintained indefinitely. Surgery of metastatic residual responding disease can be used when reasonably feasible, but its added value prognostically is unproven. When imatinib fails and/or is ineffective, other available TKIs and judicious use of surgery of limited progression are resorted to (see Table 55.1 for conventionally used agents). This treatment strategy has substantially improved the prognosis of advanced GIST patients by increasing median survival in terms of years if compared to any historical series, with a proportion of patients, limited though it may be, becoming long-term progression-free survivors.

When the disease is localized, surgery is the treatment mainstay. Indeed, all GISTs ≥2 cm in size should be resected when possible, because none of them can be considered benign. The management of GISTs <2 cm in size is more questionable.45–47 Although the low risk of progression of GISTs <2 cm leads to the recommendation of a conservative approach, a reliable mitotic index cannot be determined by biopsy or fine-needle aspiration (FNA), thus preventing the identification of those at higher risk. Therefore, both observation and resection for GISTs 1 to 2 cm can be considered, and the risks and benefits of one versus the other should be discussed with the patient. The endoscopic resection of small gastric GISTs could be an option in these presentations. Risks of perforation may be low, although the decision is made on a case-by-case basis. Regardless of their size, any small GIST that is symptomatic (e.g., bleeding from erosions through the mucosa) or increases in size on serial follow-up should be resected.
A laparotomic or laparoscopic/laparoscopy-assisted resection of primary GISTs should be performed following standard oncologic principles. On laparotomy/laparoscopy, the abdomen should be thoroughly explored to identify and remove any previously undetected peritoneal metastatic deposits.48–51 Although primary GISTs may demonstrate inflammatory adhesions to surrounding organs, true invasion is not frequent. The goal of surgery is R0 excision. A macroscopically complete resection with negative or positive microscopic margins (R0 or R1 resection, respectively) is associated with a better prognosis than a macroscopically incomplete excision (R2 excision).52,53 Available series have not clearly shown that R1 surgery is associated with a definitely higher risk of local failure, so that the decision whether to reexcise a lesion already operated on with microscopically positive margins is doubtful, aside from the fact that sometimes a reexcision may not be technically foreseeable in the gastrointestinal tract. An exception are GIST of the rectum, where microscopically positive margins are clearly associated with a higher risk of local failure.54 In general, local relapse after R0 surgery is very unlikely in GISTs. Of course, the margins of a big lesion toward the peritoneum will not be covered by any clean tissue, and this may well be the main reason for the high peritoneal relapse rate of large tumors even after complete surgery. Tumor rupture or violation of the tumor capsule during surgery are associated with a very high risk of recurrence, and therefore should be avoided.41 Some clinicians approach ruptured GISTs as already metastatic, although there may be different kinds of rupture, possibly leading to different risk levels. A lymphadenectomy is not routinely required, because lymph nodes are rarely involved (in adult patients) and are thus resected only when they are clinically suspect.
In general, surgery is a wedge or segmental resection of the involved gastric or intestinal tract, with margins that can be less wide than for an adenocarcinoma. Sometimes, a more extensive resection (e.g., total gastrectomy for a large proximal gastric GIST, pancreaticoduodenectomy for a periampullary GIST, or abdominoperineal resection for a low rectal GIST) is needed. In the rare syndromic GIST (either SDH deficient or NF-1 related), tumors are often multifocal and confined either to the stomach (SDH-deficient GIST) or the small bowel (NF-1–related GIST). The extent of surgery should be decided on a case-by-case basis, taking into account the risk of recurrence, the lack of benefit from currently available TKIs, and the actual behavior of the underlying disease.55
Adjuvant medical therapy with imatinib was demonstrated to substantially improve relapse-free intervals, although with a trend to lose the benefit in a time span of 1 to 3 years from the end of therapy.56–58 This was shown through randomized trials that compared 1 and 2 years of adjuvant therapy with imatinib versus no adjuvant therapy, and 3 years versus 1 year of adjuvant therapy with imatinib. As of today, the suggestion from these studies is that adjuvant therapy with TKIs can delay, but probably not avoid, a relapse, if this is due to occur. This correlated with a survival improvement in one trial57 and with a trend to improvement of a potential surrogate for survival in another,58 where the surrogate was survival free from changing the original tirosine kinase inhibitor (TKI)—in practice, survival without secondary resistance. In fact, secondary resistance is the limiting factor of TKIs in the advanced setting, so that an adjuvant therapy will be beneficial as long as it either avoids recurrences or at least prolongs freedom from secondary resistance, but by no means shortens it. Thus, the risk of any detrimental effect was ruled out for adjuvant therapy durations up to 3 years. In this sense, going beyond 3 years would seem logical, given the tendency to lose the benefit after 1 to 3 years from stopping adjuvant therapy, but such a policy should be validated by clinical trials ruling out any adverse effect on secondary resistance. Results from clinical studies on longer durations of adjuvant therapy are therefore expected. Currently, adjuvant therapy is recommended for 3 years and is reserved for patients with a significant risk of relapse, as long as the benefit in absolute terms will be higher as the risk increases, as is the case with all adjuvant therapies. In a sense, the lack of a tangible impact on the long-term relapse rate encourages one to exclude relatively low-risk patients, which is, to some extent, at odds with what is done with adjuvant cytotoxic chemotherapy in some solid cancers. This said, the magnitude of risk that is worth an adjuvant therapy with imatinib for 3 years may well be subject to a shared decision making with the individual patient, and, as a matter of fact, is generally placed above 30% to 50%. Logically, a benefit can be expected for patients whose genotype is potentially sensitive to imatinib.59 In practice, this leads to the selection of all patients with a KIT-mutated GIST or a PDGFRA-mutated sensitive GIST (with the exception of the D842V exon 18 mutation and the few others which are insensitive in vitro and in vivo to imatinib). Given the benefit shown with the use of a double dose of imatinib (800 mg daily) for advanced GIST patients with an exon 9 KIT-mutated GIST,60 such a dosage can be selected for them, although there is a lack of any formal demonstration in the adjuvant setting. WT GISTs are much less sensitive to imatinib, and adjuvant studies are lacking with other TKIs, which may be potentially more active. Even more importantly, the natural history of WT GISTs is often less aggressive. These are the reasons why many clinicians currently do not select WT GISTs patients for any adjuvant treatment.
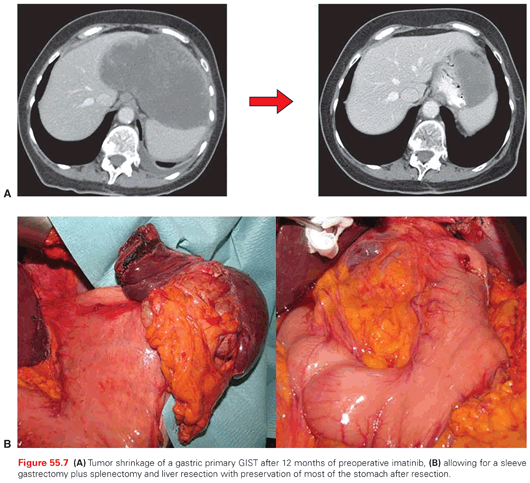
Given the extensive use of adjuvant therapy with imatinib in the high-risk populations and the activity of the drug, several recent multi-institutional retrospective series have questioned the need for extensive resections such as pancreaticoduodenectomy, abdominal perineal resection, or total/proximal gastrectomy, when tumor downsizing can be likely achieved with a preoperative medical treatment. In practice, preoperative imatinib can shrink gastric, periampullary, or rectal GISTs to such an extent as to allow more limited excisions (wedge gastrectomy, excision of periampullary lesions, transanal/perineal resection of rectal GISTs, respectively), and imatinib can then be continued postoperatively to complete the adjuvant treatment (Fig. 55.7). Thus, if extensive surgery is required for complete tumor removal, preoperative imatinib should be considered.61–64 In addition to this, there are some big abdominal masses that may be felt by the surgeon as implying a significant risk of tumor rupture during surgery, which can be treated with preoperative imatinib. Because downsizing is the clinical end point in these cases, the duration of pre-operative medical therapy is generally 6 to 12 months, which corresponds to the time interval when the maximum degree of tumor shrinkage was shown to occur in studies on advanced GISTs.65 In addition, mutational status is important in order to select patients likely to respond to imatinib, and tumor response should be monitored closely. Positron-emission tomography (PET) scans are a resource, because they can demonstrate tumor responsiveness in a matter of weeks.
Syndromic GISTs can present with multifocal and/or multicentric disease, which may imply delicate surgical decisions. Thus, in WT GISTs, such as those occurring in children and young adults or in NF-1 patients, one should take into account the indolent behavior of many lesions and the possible presence of hyperplasia of the interstitial cells of Cajal on one side, and the possibility that single lesions may be aggressive on the other. Surgery should judiciously factor in all this. In addition, the relative lack of sensitivity of WT GISTs to available TKIs may suggest to resort to surgery more liberally than is currently done with KIT-mutated GISTs.
With regard to the highly rare syndromes of familial GISTs from germ-line mutations of KIT or PDGFRA, treatment is challenging and may involve resorting to surgery and/or TKIs depending on the behavior and extent of clinically relevant lesions.
When the disease is metastatic or locally advanced, medical therapy is the best choice and is currently based on imatinib continued indefinitely.66–69
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


