- The heterogeneous and progressive nature of type 2 diabetes presents an ongoing requirement for additional and novel blood glucose-lowering agents.
- Longer-acting (once daily and once weekly injected) analogs of the incretin hormone glucagon-I ike peptide 1 (GLP-1) and new dipeptidyl peptidase 4 inhibitors are being developed.
- Proof of principle studies have shown potential for non-peptide molecules to act as GLP-1 receptor agonists and insulin mimetics.
- Potentiators of insulin action include specific phosphatase inhibitors and other types of compounds that increase insulin receptor and early post-receptor signaling.
- Selective peroxisome proliferator-activated receptor modulators, glucagon receptor antagonists and cellular glucocorticoid inhibitors have shown potential.
- Agents that directly increase glucose disposal (e.g. glucokinase activators) or reduce glucose output (e.g. fructose 1,6-bisphosphatase inhibitors) are receiving attention.
- Sodium-glucose co-transporter 2 inhibitors can facilitate glycemic control through increased renal glucose elimination.
Introduction
Current strategies for managing type 2 diabetes mellitus (T2DM) are described in Part 6. This chapter reviews new therapeutic approaches to address the metabolic disturbances of T2DM. The main focus is on drugs that have recently become available for clinical use or have entered clinical trials, and established agents that have found new applications in patients with diabetes. Some agents in preclinical development that have particular theoretical promise are also considered.
There is a continuing need for new and improved agents to treat diabetes as available therapies do not reinstate normal glucose homeostasis or eliminate the threat of long-term complications. Indeed, patients with T2DM incur chronic tissue damage and suffer premature death, even with assiduous use of the available therapies [1–3]. Lifestyle measures, notably diet and exercise, remain the foundation therapy [4], while pharmacologic interventions are added to provide further recourse against the multiple, heterogeneous and progressive endocrine and metabolic disturbances of the disease [5], Recent trial outcomes have re-emphasized the importance of comprehensive risk factor management in which early, effective, individualized and sustained glycemic control can defer the onset and reduce the severity of complications [1,2]. In particular, “glycemic memory” requires good glycemic control early after diagnosis to minimize the complications of hyperglycemia much later in the disease process [1,2,6].
Development of new antidiabetic agents
As diabetes must usually be treated for the remainder of the patient’s life, any new antidiabetic drug must be safe, well-tolerated, conveniently administered and carry minimal risk of serious hypoglycemia. It should offer durable efficacy and preferably other advantages – for example, a novel mode of action or favorable pharmacokinetics that suit a particular group such as the elderly. Ideally, a new agent will be suited to combination therapy with at least some of the existing agents, and confer benefits against conditions that are commonly associated with diabetes, such as abdominal obesity, dyslipidemia, hypertension and other vascular diseases or risk factors. A new drug might correct at least one of the major underlying endocrine or metabolic disturbances such as counter insulin resistance, improve β-cell function, reduce hyperglucagonemia or act directly to decrease glucotoxicity or lipotoxicity.
The development of a drug from a new chemical entity through to marketing approval involves many stages of rigorous preclini-cal and clinical evaluation (Table 60.1). The process can take 10–15 years and cost US$ 500–2000 million [7], If there is evidence of increased adverse cardiovascular outcomes during the pre-authorization studies, depending on the circumstances, either additional pre-authorization or post-authorization trials will be required. To extend the period of marketing exclusivity it is not unusual for companies to seek additional indications and develop new formulations post-authorization. Because many patients with T2DM require combination therapy, there is also considerable interest in the production of single-tablet fixed-dose combinations [8].
Table 60.1 Stages in the development of a new drug.
Preclinical stages New chemical entity
|
Classification of new antidiabetic agents
Blood glucose-l owering agents may be either hypoglycemic or antihyperglycemic [9]. Both reduce hyperglycemia, but hypogly-cemic agents can lower blood glucose concentrations below the euglycemic range, and therefore carry the risk of clinical hypogly-cemia. Such agents include potent inhibitors of hepatic glucose output, insulin secretagogues that act at low glucose concentrations, potent insulin-mimetic drugs and agents that impair counter-regulatory mechanisms. By contrast, antihyperglycemic agents, when used as monotherapy, do not lower blood glucose into the range of overt hypoglycemia. These drugs include the inhibitors of carbohydrate digestion and intestinal glucose absorption, anti-obesity agents, weaker suppressors of hepatic glucose output or counter-regulation, mild or glucose-dependent insulin secretagogues, most insulin-sensitizing agents and modulators of lipid metabolism.
The major glucose-lowering drugs currently used to treat diabetes, and some other agents with glucose-lowering activity, are shown in Table 60.2. Examples of other types of compounds under investigation as potential glucose-lowering drugs are listed in Table 60.3 [10,11] and illustrated in Figure 60.1.
Table 60.2 Established blood glucose-lowering agents and some other agents with blood glucose-lowering activity.
| Agents | Main mode of action |
| Agents used in the treatment of diabetes | |
| Metformin (biguanide) | Counters insulin resistance |
| Sulfonylureas | Stimulate insulin secretion |
| Prandial insulin releasers | Stimulate insulin secretion (rapid/short acting) |
| (meglitinides) | |
| Thiazolidinediones | Increase insulin sensitivity (PPARγ agonists) |
| (glitazones) | |
| GLP-1 analogs | Potentiate insulin secretion |
| DPP-4 inhibitors (gliptins) | Potentiate insulin secretion |
| a-Glucosidase inhibitors | Inhibit carbohydrate digestion |
| Pramlintide | Satiety, slow gastric emptying, decrease glucagon |
| Insulin | Decrease glucose production, increase glucose disposal, storage and utilization |
| Other agents with blood glucose-lowering activity | |
| Acipimox | Decrease lipolysis, reduce NEFAs |
| Alcohol | Inhibit gluconeogenesis |
| Niacin | Decrease lipolysis, reduce NEFAs |
| Quinine | Stimulate insulin secretion |
| Salicylates | Stimulate insulin secretion |
| γ-Adrenoceptor blockers* | Inhibit adrenergic counter-regulatory response |
DPP-4, dipeptidyl peptidase 4; GLP-1, glucagon-like peptide 1; NEFA, non-esterified fatty acids; PPARγ, peroxisome proliferator-activated receptor γ.
* β-Adrenoceptor blockers usually raise glucose concentrations, but they can contribute to hypoglycemia by inhibiting the acute counter-regulatory response and impairing hypoglycemia awareness.
Table 60.3 Examples of some new or investigational blood glucose-lowering agents.
| type of agent | action | Example |
| α-glucosidase | Inhibit carbohydrate | Voglibose |
| Inhibitors | digestion | |
| Insulin secretagogues | Stimulate insulinsecretion | Mitiglinide |
| Insulin secretion | Enhance nutrient-stimulated | GLP-1 analogs, |
| potentiators | insulin secrtion | DPP-4 inhibitors |
| Insulin mimtics | Insulin-like effects on | IGF-I |
| glucose metabolism | ||
| Insulin action | Enhance action of insulin | Selective PPAR |
| potentiators | modulators | |
| Counter-regulatory | Suppress secretion/action of | Glucagon receptor |
| hormone inhibitors | counter-regulatory | antagonists, cellular |
| hormones | glucocortocoid | |
| inhibitors | ||
| Direct glicose | Increase glucose | Gk activators, |
| regulators | metabolism | F116Pase inhibitors |
| Lipid regulators | Inhibit oxidation of fatty | CPT-1 inhibitors |
| acids | ||
| SGLTZ inhibitors | Increase renal glucose | “Flozins” |
| elimination |
CPT-1, carnitine palmitoyl transferase-1; DPP-4, dipeptidyl peptidase 4; F16Pase, fructose 1,6-bisphosphatase; GK, glucokinase; GLP-1, glucagon-like peptide-1; IGF-I, insulin-like growth factor I; PPAR, peroxisome proliferator-activated receptor; SGLT2, secondary active sodium glucose co-transporter 2.
Figure 60.1 Potential sites of action of blood glucose-lowering agents. DPP-4, dipeptidyl peptidase 4; GK, glucokinase; GLP-1, glucagon-like peptide 1; SGLT-2, sodium-glucose co-transporter 2; TZD, thiazolidinedione.
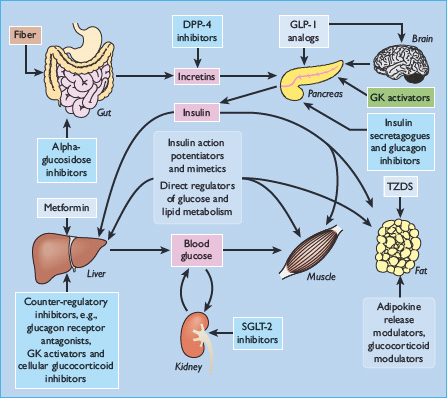
Inhibitors of intestinal carbohydrate digestion and absorption
Delaying the digestion and absorption of dietary carbohydrate in the intestine reduces post-prandial hyperglycemia and may have some “carry-over” benefit to reduce basal glycemia. Efficacy is variable but generally modest, although this approach is usually suitable for combination with most other antidiabetic therapies including insulin. Extending the period of digestion in this way can also reduce interprandial hypoglycemia in insulin-treated patients. Included here are dietary fiber supplements and inhibitors of digestive enzymes (Table 60.4).
Table 60.4 Soluble and insoluble fiber supplements.
| Soluble fiber | Insoluble fiber |
| Gums | Celluloses |
| Pectins | Wheat bran |
| Hemicelluloses | |
| Mucilages |
Dietary fiber supplements
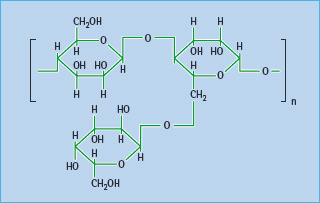
Most dietary fiber comprises plant polysaccharides that are not digested or fermented in the large bowel; some are soluble and form bulky viscous gels and gums, while others are coarse and insoluble (Table 60.4). Soluble fiber appears to be more effective than insoluble in reducing post-prandial hyperglycemia and hyperinsulinemia. Fibers can act as a barrier to diffusion within the lumen of the small intestine, where complex starchy carbohydrates are digested. Carbohydrates become entrapped within the matrix, impeding access of digestive enzymes, and restricting the diffusion of liberated saccharides across the unstirred layers of gut contents to the intestinal epithelium [12]. Various fiber supplements have appeared intermittently on the pharmacy shelves. These include common food thickeners such as soluble guar gum (E412), a galactomannan from the Indian cluster bean (Cyamopsis tetragonoloba; Figure 60.2) and fruit pectins. The prandial blood glucose-l owering effect of these supplements is usually small, especially if patients are already consuming a balanced diet containing fruit and vegetables [13].
Figure 60.2 Guar, a galactomannan polysaccharide from the Indian cluster bean, Cyamopsis tetragonoloba, used as a dietary fiber supplement.
Inhibitors of carbohydrate digestion
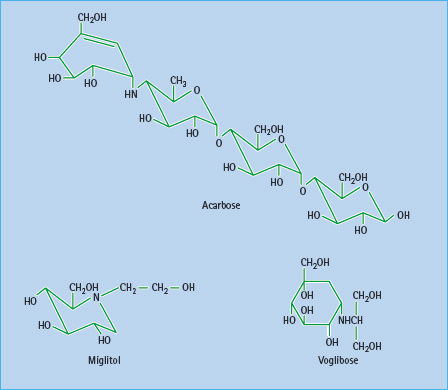
Slowing the digestion of complex carbohydrates by inhibiting amylases and glucosidases can reduce post-prandial hyperglycemia, particularly in individuals consuming a high starch diet such as rice (Figure 60.3). Inhibitors of a-amylases impair the hydrolysis of starch, but such agents have been too unpredictable for routine therapeutic use. Reversible competitive inhibitors of brush-border α-glucosidases such as acarbose, miglitol and voglibose are more predictable (Figure 60.4). These still require appropriate titration against the amount of complex carbohydrate in the diet to prevent undigested sugars from entering the large bowel, where bacterial fermentation can produce carbon dioxide and osmotically active glucose, causing flatulence and diarrhea as side effects [14]. There do not appear to be new α-glucosidase inhibitors in clinical development.
Figure 60.3 Starch digestion and its inhibitors. α(1–6)-glucosidic linkages are cleaved by α(1–6) glucosidases, and α(1–4) linkages by α-amylase. α-Glucosidase inhibitors such as acarbose act by competitive, reversible inhibition of intestinal brush-border α-glucosidase enzymes. This slows the rate of carbohydrate digestion.
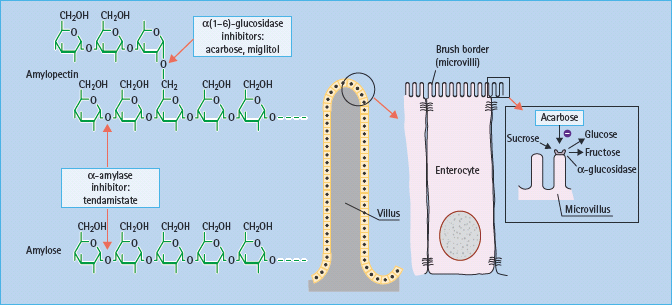
Figure 60.4 α-Glucosidase inhibitors in present use are acarbose and miglitol. Voglibose is available in some countries. These agents have different affinities for specific α-glucosidases: the binding affinity of acarbose is glucoamylase > sucrase > maltase > dextrinase; miglitol and voglibose potently inhibit sucrase, while voglibose shows more potent inhibition of other α-glucosidases than acarbose. Acarbose also weakly inhibits α-amylase.
Insulin secretagogues
Several defects in islet β-cell function occur early in the pathogenesis of T2DM: the acute first-phase insulin secretory response to glucose becomes reduced and eventually lost, processing of proinsulin to insulin is impaired, the normal pulsatile rhythm of basal insulin secretion is disturbed, and the second phase of insulin secretion is often extended, albeit diminished in magnitude, as the period of post-prandial hyperglycemia is prolonged (see Chapter 10). In advanced stages of T2DM, β-cell mass and insulin biosynthesis are also compromised. An ideal insulin secretagogue would restore β-cell sensitivity to glucose, and support adequate biosynthesis, processing and secretion of insulin in response to other nutrients, hormones and neural factors. Insulin secretagogues can be categorized into initiators (e.g. sulfonylureas), which stimulate insulin secretion on their own or require only low glucose concentrations, and potentiators (e.g. GLP-1 analogs), which enhance the effect of rising glucose and other secretagogues but do not induce insulin release without glucose.
Initiators of insulin secretion
Currently available sulfonylureas and meglitinides are oral insulin secretagogues that initiate insulin secretion (see Chapter 29). They bind to either the sulfonylurea site or the benzamide site (meglitinides) on the sulfonylurea receptor 1 (SUR1) in the plasma membrane of the β-cell. The SUR1 is part of the ATP-sensitive potassium channel (KATP channel), which consists of an octameric complex of four Kir6.2 pores (inwardly rectifying potassium channels) surrounded by four SUR1 molecules (Figure 60.5). Binding of ligands to the sulfonylurea or benzamide sites on SUR1 elicits the same response as binding of ATP to the nucleotide-binding domains of the channel, namely closure of the Kir6.2 pore [15]. This prevents K+ efflux, which leads to localized depolarization of the plasma membrane. In turn, this opens voltage-gated (L-type) calcium channels, allowing influx of Ca2+ ions, which increases the cytosolic calcium ion concentration. Calcium-sensitive proteins are thereby activated, triggering the exocytosis of insulin-containing secretory granules (Figure 60.6). SUR1–Kir6.2 channels have also been identified within the membranes of mitochondria and possibly other organelles, suggesting that sulfonylureas and meglitinides could act at these and other sites in the cell as part of their insulin-releasing action.
Figure 60.5 The ATP-sensitive (KATP ) channel in the β-cell membrane consists of a large sulfonylurea 1 (SUR1) subunit (17 transmembrane domains) and a smaller pore-forming unit that acts as an inwardly rectifying K channel, Kir6.2. The cytosolic surface of the SUR1 subunit has separate binding sites for sulfonylureas (S), benzamido compounds (B) and the nucleotides, ADP and ATP (N). Four complete channel units self-associate to form an octameric complex in the membrane.
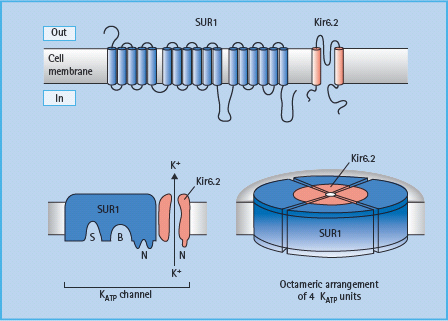
Figure 60.6 Sulfonylureas and benzamido compounds stimulate insulin release mainly by binding to sites on the sulfonylurea receptor 1 (SUR1) of the adenosine triphosphate (ATP)-sensitive potassium channels (KATP channels). This closes the Kir6.2 pores of the KATP channels, causing localized depolarization, opening of voltage-dependent calcium channels, increasing Ca2+ influx, and activation of calcium-dependent proteins that promote exocytosis of insulin granules. Imidazoline derivatives can act on I1 and I2 receptors, as well as other (unclassified) receptors and directly on the Kir6.2 pores to enhance insulin secretion. Succinate esters fuel glucose metabolism.
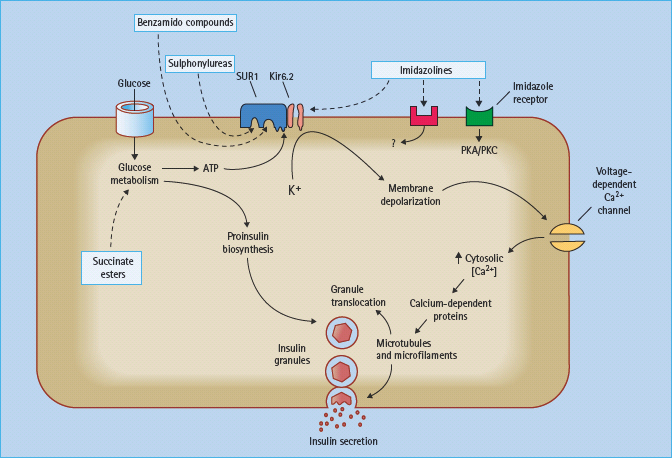
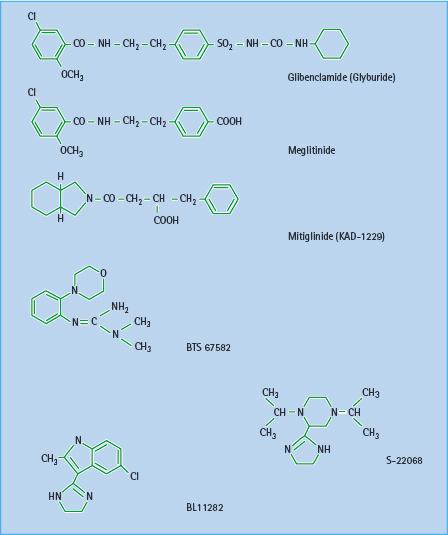
Several novel insulin secretagogues have been reported to act by closing the KATP channels. The meglitinide derivative mitiglinide (KAD-1229) appears to bind at the benzamide site on SUR1 (Figure 60.6), and this agent has proceeded in clinical development [16]. Several other types of compounds have been shown to interact with KATP channels, but these do not appear to have proceeded beyond early clinical studies (Figure 60.7). These include the morpholinoguanidine BTS67582 [17] and certain imidazolines such as S22068 [18]. Other imidazoline compounds, which bind to I1, I2 and probably other receptors, may also close KATP channels as part of their mechanisms to stimulate insulin secretion [19]; however, there are imidazolines such as BL11282 that stimulate glucose-induced (but not basal) insulin secretion by effects on protein kinases without closing KATP channels [20]. Various α2-adrenergic receptor antagonists such as phentolamine, which can reduce the tonic suppression of insulin secretion mediated through α2-adrenergic activation, may also act to close KATP channels.
Figure 60.7 Structures of some established and novel insulin secretagogues. Glibenclamide (glyburide) has both a sulfonylurea and a benzamide moiety. Meglitinide derivatives exhibit a benzamide moiety. BTS 67582 is a morpholinoguanidine, while S-22068 and BL11282 are examples of imidazoline compounds.
Although closure of KATP channels initiates insulin secretion, it does not promote insulin biosynthesis. Agents that increase nutrient metabolism, and close KATP channels through increased ATP production, additionally enhance proinsulin biosynthesis. This capability is illustrated by esters of succinic acid which provide a metabolisable substrate to the mitochondria [21]. These compounds have low enteral bioavailability and a short duration of action, and they also fuel gluconeogenesis in the liver. A related approach could be to stimulate mitochondrial succinylcoenzyme A (CoA) synthetase to generate ATP and guanosine triphosphate (GTP) [22].
Because the islet β-cell takes up glucose approximately in proportion to the circulating glucose concentration and phosphorylates the glucose via glucokinase (GK; EC 2.7.1.1), the β-cell is amenable to stimulation of glycolysis with agents that enhance the activity of GK [23]. Although this effect is actually potentiating the action of glucose (Figure 60.8), it can operate at low glucose concentrations, so it has been categorized here as an initiator. Several specific small molecule activators of GK have been shown to increase insulin secretion and improve glucose homeostasis in models of T2DM. These include allosteric activators and molecules that prevent association of GK with or cause dissociation of GK from inhibitory protein complexes [24–29]. Several of these agents are now in clinical trial, and it will be interesting to see if this approach can benefit other aspects of islet function and be regulated to avoid hypoglycemia. Because liver cells express GK and take up glucose approximately in proportion to the circulating concentration, GK activators will also stimulate hepatic glucose utilization and reduce hepatic glucose production, adding to their blood glucose-lowering potency.
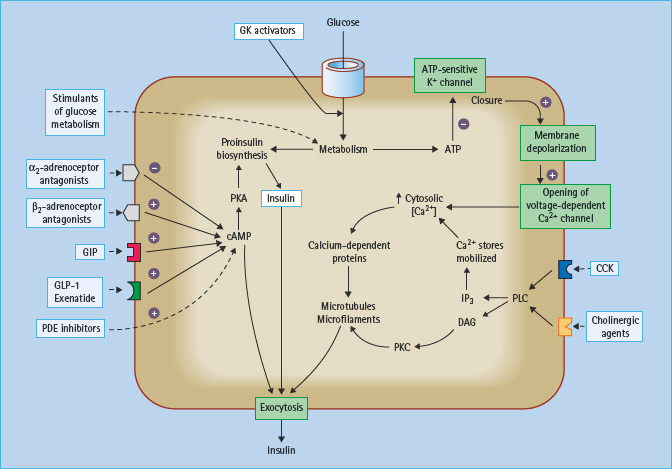
Figure 60.8 Sites on the pancreatic β-cell that offer potential targets to potentiate nutrient-induced insulin secretion. cAMP, cyclic adenosine monophosphate; CCK, cholecystokinin; DAG, diacylglycerol; GIP, gastric inhibitory polypeptide, also known as glucose-dependent insulinotropic peptide; GK, glucokinase; IP3, inositol-1,4,5-trisphosphate; PDE, phosphodiesterase; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C. Activated ated β2-adrenoceptors and GIP and glucagon-like peptide-1 (GLP-1) receptors stimulate insulin secretion, as do antagonists of the inhibitory α2-adrenoceptors.
Potentiators of insulin secretion
Potential opportunities to increase nutrient-induced insulin secretion are shown in Figure 60.8. In principle, these agents should predominantly decrease post-prandial hyperglycemia and carry less risk of interprandial hypoglycemia.
Incretins
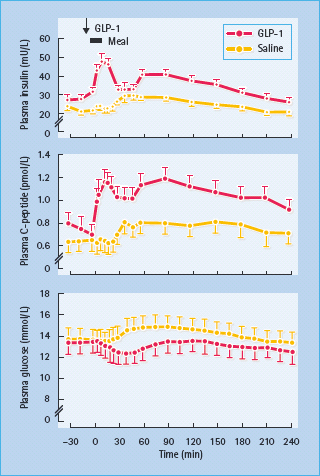
Insulin secretion is enhanced by several hormones released from the gut during feeding – so-called incretin hormones. The main incretins are glucagon-like peptide 1 (7–36) amide (GLP-1) and gastric inhibitory polypeptide (GIP; also known as glucose-dependent insulinotropic peptide). These hormones activate specific G-protein coupled receptors in the β-cell membrane and potentiate nutrient-induced insulin secretion and insulin biosynthesis, at least partly through increased cyclic adenosine monophosphate (cAMP) and production of protein kinase A (PKA) [30,31]. Both hormones have also been shown to promote β-cell mass in animal models, possibly slowing β-cell apoptosis and increasing β-cell neogenesis by increased expression of the transcription factor PDX-1 (pancreatic duodenal homeobox 1), which promotes proliferation and differentiation of ductal progenitor cells (Figure 60.9). GLP-1 has been favored as a treatment for T2DM because it also suppresses glucagon secretion in a glucose-dependent manner, slows gastric empting and exerts a satiety effect that can facilitate weight loss [32]. Moreover, GLP;-1 concentrations appear to be reduced in T2DM but the ability of the peptide to enhance insulin release is maintained (Table 60.5). Thus, when injected subcutaneously immediately before a meal, GLP-1 can substantially reduce the post-prandial rise in blood glucose concentrations in T2DM (Figure 60.10). However, the rapid degradation of GLP-1, mostly by the enzyme dipeptidyl peptidase 4 (DPP-4) and the renal clearance of GLP-1 (plasma half-life < 2 minutes) makes the peptide itself an inconvenient form of therapy [33].
Figure 60.9 Incretin hormones GLP-1 and GIP have been shown to increase β-cell mass in animal models. Possible mechanisms include increased β-cell proliferation and differentiation, slowing of β-cell apoptosis and increased β-cell neogenesis from ductal progenitor cells.
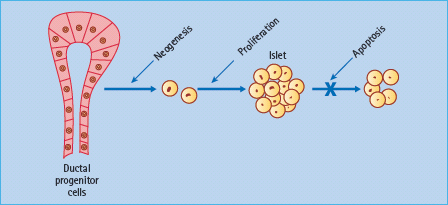
Table 60.5 Effects of the incretin hormones glucagon-like peptide 1 (7–36) amide ( GLP-1) and gastric inhibitory polypeptide ( GIP ; also known as glucose-dependent insulinotropic peptide).
| GIP | GLP-1 | |
| Pantic | ||
| Glucose-induced | Increase | Increase |
| insulin secretion | ||
| Prainsulin bisynthesis | Increase | Increase |
| β-Cell mass(rodent) | Increase | Increase |
| Glucagon secretion | Increase or no effects | Decrease |
| Other action | ||
| Gastric emptying | No/slight effect | Decrease |
| Appetite/feeding | No significant effect | Decrease |
| Weight gain | No effect or increase | Decrease |
| Myocardial metabolism | No established effect | Possible benifts |
| Type 2 diabetes | ||
| Plasma concentrations | Normal or slight reduced | Reduced late phase |
| Insulin-releasing effect | Reduced | Mostly retained |
Figure 60.10 Insulin-releasing and glucose-lowering effects of glucagon-like peptide 1 (GLP-1) in eight obese patients with T2DM. A dose of 25 mmol was injected subcutaneously into the gluteal region 5 minutes before a standard meal. The plasma insulin and C-peptide responses were significantly greater, and the plasma glucose response significantly flatter (all P < 0.001), compared with a control (saline) injection. Reproduced from Glutniak et al. Diabetes Care 1994; 17:1039 –1044, with permission from the American Diabetes Association.
DPP-4 cleaves off an N-terminal dipeptide where there is an alanine residue (as in GLP-1) or a proline residue at the N2 position. Exenatide (exendin-4) is a GLP-1 analog with a glycine residue at the N2 position (Figure 60.11), making it resistant to degradation by DPP-4, and extending the half-life to several hours. Because exenatide retains the glucose-lowering efficacy of GLP-1 it has become established as an incretin therapy, injected subcutaneously twice daily before the main meals [34]. The glucose-dependent nature of the insulin-releasing and glucagon-suppressing effects is reflected in the limited risk of hypoglycemia, while the satiety effect has assisted weight loss and favored use in obese patients.
Figure 60.11 The amino acid structure of GLP-1, and the GLP-1 analogs exenatide and liraglutide. GIP and glucagon are shown for comparison. DPP-4 cleaves off an N-terminal dipeptide where there is an alanine residue (as in GLP-1 and GIP) or a proline residue at the N2 position. Liraglutide is protected from degradation by DPP-4 when the palmitoyl fatty acid moiety binds to albumin.
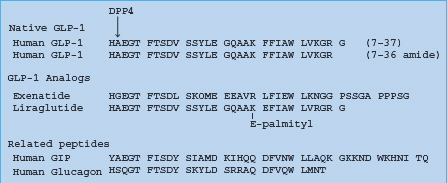
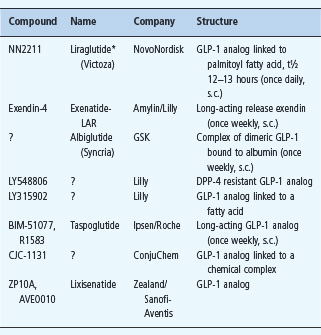
Among other GLP-1 analogs (Table 60.6), liraglutide (NN2211) recognized regulatory approval in Europe in July 2009. Liraglutide is GLP;-1 (7–37) with Lys34 replaced by Arg34, and Lys26 attached via a glutamate residue to a C16 hexadecanoyl (palmitoyl) fatty acid chain. The fatty acid chain facilitates association into heptam-ers and attachment of the molecule to albumin, protecting it from degradation by DPP-4 and enabling once daily injection [35].
Table 60.6 Peptide glucagon-like peptide 1 ( GLP-1) agonists in clinical development.
Other GLP-1 preparations in development include GLP1-INT (GLP-1 analog; Transition Therap/NovoNordisk) and MKC253 (GLP-1-technospheres for inhalation, MannKind Corp).
*Liraglutide was launched in Europe in July 2009.
A once-weekly depot formulation of exenatide, new long-acting GLP-1 analogs, slow-release formulations and different administration routes (e.g. transdermal, buccal and inhaled) are receiving clinical assessment [32]. Hybrid peptides are also being explored to increase glucose-lowering efficacy, for example DAPD, a peptide that is a GLP-1 receptor agonist and a glucagon receptor antagonist [36]. To circumvent the need for injections, non-peptide GLP-1 receptor agonists (e.g. Boc5) have been identified. These bind to the GLP-1 receptor on islet β-cells and enhance glucose-dependent insulin secretion [37,38].
Although GIP potentiates nutrient-induced insulin secretion it also increases glucagon release and promotes lipid deposition [39]. Moreover, GIP receptor knockout (KO) mice and the administration of GIP receptor antagonists (both peptide and non-peptide) have been shown to prevent the development of obesity, improve glucose homeostasis and reduce insulin resistance in animal models [40–44]. This is consistent with evidence from bariatric surgery: a reduced supply of nutrients through the proximal small intestine (location of GIP-secreting K-cells) rapidly improves glycemic control in obese patients with diabetes [45]. Thus, GIP antagonism by pharmacologic means or “metabolic surgery” may provide a new antidiabetic mechanism [46].
DPP-4 inhibitors
Inhibiting the enzyme DPP-4 (EC 3.4.14.5) prevents the rapid inactivation of the endogenous incretins GLP-1 and GIP, thereby raising their circulating concentrations and increasing nutrient-stimulated insulin release and other effects of these incretins (Table 60.5). DPP-4 is found “loose” in the circulation and tethered to cell membranes, especially endothelia in capillaries of the gastrointestinal tract, a location that facilitates the degradation of incretins. The protease activity of DPP-4 degrades a range of biologically active peptides in addition to incretins, including substance P, bradykinin, peptide YY, neuropeptide Y, pituitary adenylate cyclase-activating peptide, insulin-l ike growth factor I (IGF-I) and various interleukins and monocyte chemo-attractant proteins [33,47]. Despite affecting this wide range of peptides, to date, specific DPβ-4 inhibitors have not shown significant adverse effects during substantial clinical use [32]. Also, DPP-4 is the lymphocyte cell surface protein CD26 required for the co-stimulation response to recall antigens, but its immunologic role does not appear to be interrupted by small molecule inhibitors of its peptidase activity.
Currently available DPP-4 inhibitors (gliptins) include sitagliptin, vildagliptin and saxagliptin (see Chapter 30). These agents improve glycemic control similarly to GLP-1 analogs but there are subtle differences that may partly reflect the concomitantly increased GIP, and the likelihood that raised endogenous incretin concentrations will not achieve the high concentrations of exogenously administered GLP-1 analogs. In consequence, DPP-4 inhibitors are unlikely to cause initial nausea through delayed gastric emptying, and they may exert a lesser satiety effect resulting in little change of body weight. Among the DPP-4 inhibitors in clinical development (Table 60.7), trials with alogliptin are well advanced (Figure 60.12). These agents have shown high specificity for DPP-4 inhibition, and preliminary information indicates that therapeutic concentrations cause almost complete inhibition of DPP-4 activity for about 12 hours, producing similar glucose-lowering efficacy to existing gliptins [32].
Table 60.7 DPP-4 inhibitors recently available and in clinical development.
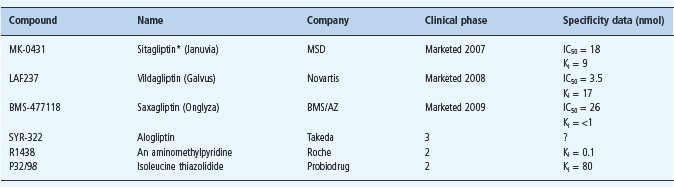
AZ, AstraZeneca; BMS, Bristol Myers Squibb; MSD, Merck Sharp Dohme; ?, not reported.
Other compounds under consideration include PSN9301 (Prosidion), GR-8200 (Glenmark), PHX-1149 (Phenomix), SSR-162369 (Sanofi-Aventis), ALS 2-0426 (Alantos/ Amgen), and NN-7201 (NovoNordisk).
*Launched in Mexico in 2006, and in USA, UK and several other European countries in 2007.
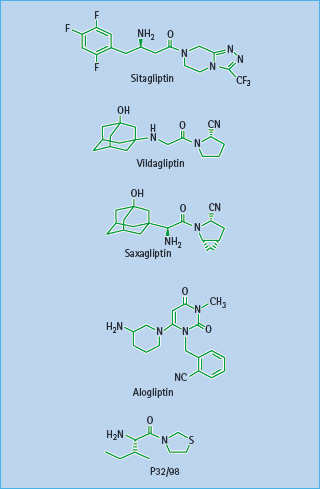
Figure 60.12 Structures of the DPP-4 inhibitors sitagliptin, vildagliptin, saxagliptin, alogliptin and P32/98.
Phosphodiesterase inhibitors and other approaches
The β-cell expresses several phosphodiesterases (PDEs) that degrade cAMP and so reduce insulin release. Selective and transient inhibition of these enzymes in β-cells, especially isoform PDE-3B, which exerts most influence on glucose-induced insulin secretion, could be a possible intervention [48], but the problem of specifically targeting the β-cells has yet to be overcome. Other theoretical approaches to potentiate insulin secretion (Figure 60.8), such as antagonism of α2-adrenoceptors and activators of phospholipase C (PLC), have not been possible to target specifically at the β-cell.
Insulin-mimetic drugs
Insulin resistance (impaired insulin action) is a typical feature of T2DM, but it is highly heterogeneous, initially progressive (excepting some monogenic forms), and susceptible to many different genetic and environmental factors [49,50]. Most patients with T2DM probably incur multiple defects that impinge on insulin receptor function and/or various post-receptor signaling pathways, as well as independent disturbances in the activities of substrate transporters and metabolic enzymes consequent to glu-cotoxicity and lipotoxicity [51]. Indeed, insulin has important genomic effects that determine the expression levels of many cellular components that are directly and indirectly involved in metabolic homeostasis. Defects of insulin receptor structure are uncommon, and reductions in insulin receptor number are not usually rate limiting. Thus, the therapeutic challenge of insulin resistance appears to require interventions at diverse intracellular targets [52].
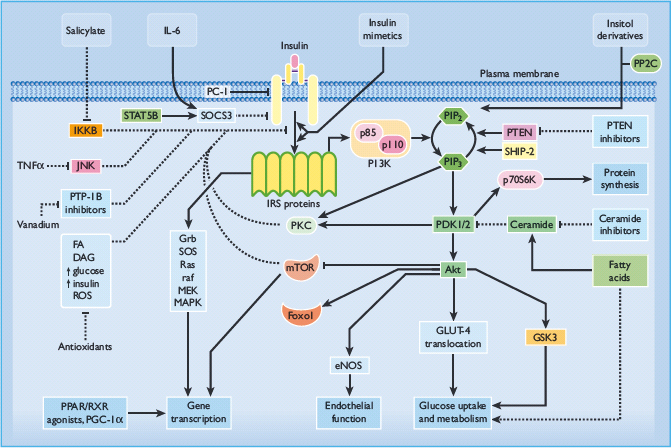
In theory, agents that address defects of insulin receptor signaling or early post-receptor lesions might be expected to produce a broader spectrum of benefits, but if the rate-limiting defects occur at more distal locations their therapeutic efficacy will be compromised. Potential target sites to obviate cellular defects of insulin action are shown in Figure 60.13.
Figure 60.13 Pathways of intracellular insulin signaling showing some of the potential sites for therapeutic intervention. Adapted from Bailey [52]. Akt, protein kinase B (PKB); AMPK, adenosine monophosphate-activated protein kinase; DAG, diacylglycerol; eNOS, endothelial nitric oxide synthase; FA, fatty acid; FOXO1, forkhead box protein O1A; GLUT-4, glucose transporter 4; Grb, growth factor receptor binding protein; GSK3, glycogen synthase kinase 3; IKKB, inhibitor κ-B kinase-β ; IL-6, interleukin 6; IRS, insulin receptor substrate; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase kinase; mTOR, mammalian target of rapamycin; PC-1/NNP1, glycoprotein-1; PDK, phosphoinositide-dependent protein kinase; PGC-1 α, PPAR coactivator 1α; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol-3,4-bisphosphate; PIP3, phosphatidylinositol-3,4,5-trisphosphate; PKC, protein kinase C; PPAR, peroxisome proliferator-activated receptor; PP2C, pyruvate dehydrogenase phosphatase (protein phosphatase 2C); PTEN, protein phosphatase PTEN; PTP-1B, protein tyrosine phosphatase-1B; Raf, a serine-threonine protein kinase; Ras, a guanosine triphosphatase; ROS, reactive oxygen species; RXR, retinoid X receptor; SHIP-2, src homology-2-inositol phosphatase; SOCS-3, suppressor of cytokine signaling-3; SOS, sons of sevenless; STAT, signal transducer and activator of transcription; TNF-α, tumour necrosis factor α; ↑, increase; →, stimulatory effect; ⊣, inhibitory effect.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


