INTRODUCTION
SUMMARY
Much of our immune defense against invading organisms is predicated upon the tremendous diversity of immunoglobulin molecules. Immunoglobulins are glycoproteins produced by B lymphocytes and plasma cells. These molecules can be considered receptors because the primary function of the immunoglobulin molecule is to bind antigen. A single person can synthesize 10 to 100 million different immunoglobulin molecules, each having a distinct antigen-binding specificity. The great diversity in this so-called humoral immune system allows us to generate antibodies specific for a variety of substances, including synthetic molecules not naturally present in our environment. Despite the diversity in the specificities of antibody molecules, the binding of antibody to antigen initiates a limited series of biologically important effector functions, such as complement activation and/or adherence of the immune complex to receptors on leukocytes. The eventual outcome is the clearance and degradation of the foreign substance. This chapter describes the structure of immunoglobulins and outlines the mechanisms by which B cells produce molecules of such tremendous diversity with defined effector functions.
Acronyms and Abbreviations
ADCC, antibody-dependent cellular cytotoxicity; AID, activation-induced deaminase; BACH2, basic leucine zipper transcription factor 2; BCL-6, B-cell chronic lymphocytic leukemia/lymphoma 6; BiP, immunoglobulin-binding protein; Blimp-1, B-lymphocyte-induced maturation protein-1; BLNK, B-cell linker protein; BTK, Bruton tyrosine kinase; C, constant; CDR, complementarity determining region; CRI, cross-reactive idiotype; CSR, class switch recombination; D, diversity; DLBCL, diffuse large B-cell lymphoma; DNA-PK, DNA protein kinase; E2F1, E2F transcription factor 1; EBF1, early B-cell factor 1; ERGIC, ER-Golgi-intermediate compartment; FR, framework region; H, heavy; HMG, high-mobility group protein; Ig, immunoglobulin; IL, interleukin; IRF4, interferon regulatory factor 4; ITAM, immunoreceptor tyrosine-based activation motif; κ, immunoglobulin kappa light chain; Kde, kappa-deleting element; λ, immunoglobulin lambda light chain; L, light; MITF, microphthalmia-associated transcription factor; MYBL1 and 2, v-myb myeloblastosis viral oncogene homologues 1 and 2; NHEJ, nonhomologous DNA end-joining; PAX5, paired box gene 5; PDI, protein disulphide isomerase; PLC, phospholipase C; POU2AF1, Pou domain, class 2, associating factor 1; POU2F2, Pou domain, class 2, factor 2; PRDM1, positive regulatory domain 1-binding factor-1; RAG, recombination-activating gene; RSS, recombination signal sequence; SCID, severe combined immunodeficiency; SHP-1, Src homology 2 domain-containing protein tyrosine phosphatase-1; SHIP-1, phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase 1; TCFE2A, transcription factor E2a; UNG, uracil-DNA glycosylase; V, variable-region gene; V(D)J, exon created by a rearranged immunoglobulin heavy-chain variable-region gene, diversity gene segment, and joining gene segment; XBP1, X-box binding protein-1.
IMMUNOGLOBULIN STRUCTURE AND FUNCTION
All naturally occurring immunoglobulin molecules are composed of one or several basic units consisting of two identical heavy (H) chains and two identical light (L) chains (Fig. 75–1).1 The four polypeptides are held in a symmetrical, Y-shaped structure by disulfide bonds and noncovalent interactions.2,3,4 The internal disulfide bonds of the heavy and light chains cause the polypeptides to fold into compact globe-shaped regions called domains, each containing approximately 110 to 120 amino acid residues. Each domain is composed of β-pleated sheets that are stabilized by a conserved disulfide bond (Fig. 75–1). The light chains have two domains; the heavy chains have four or five domains. The aminoterminal domains of the heavy and light chains are designated the variable (V) regions because their primary structure varies markedly among different immunoglobulin molecules. The carboxy-terminal domains are referred to as constant (C) regions because their primary structure is the same among immunoglobulins of the same class or subclass. The amino acids in the light- and heavy-chain variable regions interact to form an antigen-binding site. Each four-chain immunoglobulin basic unit has two identical binding sites. The constant-region domains of the heavy and light chains provide stability for the immunoglobulin molecule. The heavy-chain constant regions also mediate the specific effector functions of the different immunoglobulin classes (Table 75–1).
| IgG | IgA | IgM | IgD | IgE | |
|---|---|---|---|---|---|
| Heavy-chain class | γ | α | μ | δ | ε |
| Heavy chain subclass | γ1, γ2, γ3, γ4 | α1, α2 | — | — | — |
| No. of heavy-chain domains | 4 | 4 | 5 | 4 | 5 |
| Secretory form | Monomer | Monomer, dimer | Pentamer | Monomer | Monomer |
| Molecular mass (Da) | 150,000 | 160,000 (monomer) 400,000 (secretory) | 900,000 | 184,000 | 188,000 |
| Antigen-binding valency | 2 | 2 (monomer) 4 (secretory) | 10 | 2 | 2 |
| Serum concentration (mg/mL) | 8–16 | 1.4–4.0 | 0.5–2.0 | 0–0.4 | 17–450 ng/mL |
| Percent of total immunoglobulin | 80 | 13 | 6 | 1 | 0.002 |
| Electrophoretic mobility | γ | Fast γ to β | Slow γ | Fast γ | Fast γ |
| Percent carbohydrate | 3 | 8 | 12 | 13 | 12 |
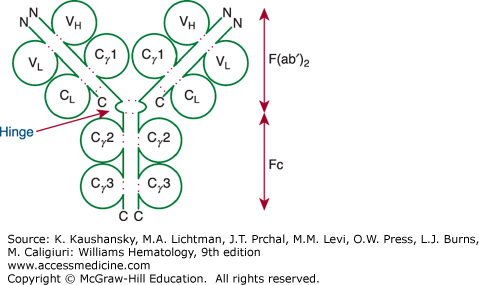
Figure 75–1.
Model of an immunoglobulin (Ig) G molecule. The light-chain domains VL and CL and the heavy-chain domains VH, Cγ1(or CH1), Cγ2 (or CH2), and Cγ3 (or CH3) are labeled inside the respective immunoglobulin domain. Dotted red colored lines indicate intrachain and interchain disulfide bonds. The aminoterminus (N) and carboxyl-terminus (C) of each polypeptide are indicated. The hinge region also is indicated. Digestion by pepsin cleaves the molecule at the carboxyl side of the hinge region, which generates Fc and F(ab′)2 fragments, as indicated on the right. The F(ab′)2 fragment is bivalent, as it is held together by the disulfide bridges in the hinge region. On the other hand, digestion of the molecule by papain degrades the Fc portion and generates monovalent Fab fragments, as the cleavage site for papain is on the aminoterminal side of the disulfide bridges of the hinge region.
Immunoglobulin light chains have an approximate Mr of 23,000. They are divided into two types, κ and λ, based upon multiple amino acid sequence differences in the single constant-region domain.5 The λ chains are divided further into subclasses. The proportion of κ-to-λ chains in adult human plasma is approximately 2:1. The immunoglobulin light-chain constant region has no known effector function. Its main purpose may be to allow for proper assembly and release of an intact immunoglobulin molecule. Soon after synthesis, the antibody light-chain constant region associates with the nascent immunoglobulin heavy chain (see Fig. 75–1), releasing the latter from the immunoglobulin-binding protein (BiP). BiP is a heat shock protein that, in the absence of antibody light chain, binds the first constant-region domain of the newly synthesized heavy chain, thereby retaining the heavy-chain polypeptide in the cell’s endoplasmic reticulum.6
Immunoglobulin heavy chains have an Mr of 50,000 to 70,000, depending upon the number and length of the constant-region domains. The five major isotypes of heavy chains—γ, α, μ, δ, and ε—determine the five corresponding classes of immunoglobulin (Ig): IgG, IgA, IgM, IgD, and IgE. The individual immunoglobulin molecules of each isotype may contain either κ or λ light chains, but not both. Tables 75–1 and 75–2 summarize the distinct physical and functional properties of the human immunoglobulin classes.
| IgG | IgA | IgM | IgD | IgE | |
|---|---|---|---|---|---|
| Percent of body pool in intravascular space | 45 | 42 | 76 | 75 | 51 |
| Percent of intravascular pool catabolized per day | 6.7 | 25 | 18 | 37 | 89 |
| Normal synthetic rate (mg/kg per day) | 33 | 24 | 6.7 | 0.4 | 0.02 |
| Serum half-life (days) | 21 | 5.8 | 10 | 2.8 | 2.3 |
| Placental transfer | Yes | No | No | No | No |
| Cytophilic for mast cells and basophils | No | No | No | No | Yes |
| Binding to macrophages and other phagocytes | Yes | No | No | No | Yes |
| Reactivity with staphylococcal protein A | Yes | No | No | No | No |
| Antibody-dependent cell-mediated cytotoxicity | Yes | No | No | No | No |
| Complement fixation | |||||
| Classic pathway | Yes | No | Yes | No | No |
| Alternative pathway | No | Yes | No | No | No |
Approximately 80 percent of the immunoglobulins in adult plasma are IgG. The IgG molecule is composed of the basic 150-kDa immunoglobulin four-chain structure plus approximately 3 percent carbohydrate. Near the junction of the two arms of the Y-shaped immunoglobulin molecule, the two heavy chains interact to form a flexible “hinge” region (see Fig. 75–1). Exposed between constant-region globular domains, the hinge region is attacked readily by the proteolytic enzyme papain or pepsin. Figure 75–1 shows the cleavage sites. Digestion of IgG with papain yields three fragments. The single Fc piece contains the carboxy-terminal region of both heavy chains. The two identical F(ab) pieces contain the entire light chain and the aminoterminal portion of the heavy chain. The Fc-regions also contain a binding epitope for the neonatal Fc receptor (FcRn), responsible for the extended half-life, placental transport, and bidirectional transport of IgG to mucosal surfaces.7 As such, IgG molecules effectively penetrate extravascular spaces and readily cross the placental barrier to provide passive immunity to the newborn.
IgG is the predominant antibody produced during the secondary immune response to antigen. The average half-life of circulating IgG molecules is approximately 21 days, although the exact value varies among the IgG subclasses (Table 75–3). Within the IgG class are four major subclasses, designated IgG1, IgG2, IgG3, and IgG4.7 The most abundant subclass is IgG1, which constitutes 60 percent of the total IgG in plasma. All IgG subclasses have a similar molecular mass except for IgG3, which has a much longer hinge region than any other IgG subclass. The IgG3 hinge region is approximately four times as long as the IgG1 hinge, containing up to 62 amino acids (including 21 prolines and 11 cysteines), forming a polyproline helix with limited flexibility. Because of this, IgG3 myeloma protein may aggregate spontaneously to produce a hyperviscosity syndrome.
| IgG1 | IgG2 | IgG3 | IgG4 | |
|---|---|---|---|---|
| Heavy chain subclass | γ1 | γ2 | γ3 | γ4 |
| Molecular mass (kDa) | 146 | 146 | 170 | 146 |
| Serum concentration (mg/mL) | 7 | 4 | 0.5 | 0.6 |
| Relative abundance (%) | 60 | 32 | 4 | 4 |
| Serum half-life (days) | 21 | 21 | 7-21a | 21 |
| Placental Transfer | ++++ | ++ | ++/++++* | +++ |
| Complement fixation (C1q binding) | ++ | + | +++ | – |
| FcR Binding | ||||
| FcγRI (CD64) | +++ | – | ++++ | ++ |
| FcγRIIaH131 (CD32)† | +++ | ++ | ++++ | ++ |
| FcγRIIaR131 (CD32)† | +++ | + | ++++ | ++ |
| FcγRIIb/c (CD32) | + | – | ++ | + |
| FcγRIIIaF158 (CD16)‡ | ++ | – | ++++ | – |
| FcγRIIIaV158 (CD16)‡ | +++ | + | ++++ | – |
| FcγRIIIb (CD16) | +++ | – | ++++ | ++ |
| FcγRn (at pH <6.5) | +++ | – | ++++ | – |
| Antibody-dependent cell-mediated cytotoxicity | + | – | + | – |
| Heterologous skin sensitization | + | – | + | + |
Each subclass has a distinct heavy-chain constant region and mediates different effector functions (see Table 75–3). Whereas IgG1 and IgG3 proteins activate complement via the classic pathway, IgG2 molecules fix complement poorly and IgG4 proteins not at all.
Antibody responses to soluble protein antigens and membrane proteins primarily induce IgG1, but also lower levels of the other subclasses, mostly IgG3 and IgG4. On the other hand, IgG antibody responses to bacterial capsular polysaccharide antigens typically are restricted to IgG2; IgG2 deficiency can result in the virtual absence of IgG anticarbohydrate antibodies. Viral infections generally induce IgG antibodies of the IgG1 and IgG3 subclasses, with IgG3 antibodies appearing first in the course of the infection. The IgG4 subclass typically is produced in response to allergens. Because IgG4 has relatively low affinity to activating FcγRIII, but relatively high affinity to the inhibiting FcγRII, it may serve to prevent excessive immune responses against sterile antigens, such as bee venom, which do not pose infectious threats. As such, IgG4 has been called a “blocking antibody” in the context of allergy, where it may compete with IgE for allergen binding.
For antigens found on pathogens, the bound IgG can: (1) tag the pathogen for ingestion and destruction by phagocytes, a process called opsonization; (2) activate complement; and/or (3) direct antibody-dependent cell-mediated cytotoxicity (ADCC). Either aggregated IgG or antigen–antibody complexes may bind to specific receptors for the Fc fragment, designated FcγRI (CD64), FcγRII (CD32), and FcγRIII (CD16). Of the IgG subclasses, IgG1 binds best to FcγRI (CD64) and FcγRII (CD32), with affinities (dissociation constant [Kd]) of 10 nM and 50 μM, respectively (see Table 75–3). IgG1 and IgG3 bind equally well to FcγRIII (CD16), with a Kd of 2 μM (see Table 75–3). This is the Fc receptor expressed by natural killer (NK) cells (or K cells), which mediate ADCC. Proteins of the IgG4 or IgG2 subclass bind poorly to FcγRI (CD64) and bind not at all to FcγRIII (CD16) (see Table 75–3). IgG1 is the most proficient subclass at directing ADCC. For this reason, most of the therapeutic monoclonal antibodies are of the IgG1 subclass, which can be modified further to enhance their capacity to direct ADCC.8
IgA composes only approximately 13 percent of plasma immunoglobulin (see Table 75–1), even though the production of IgA exceeds that of any other immunoglobulin isotype, accounting for 60 to 70 percent of antibodies produced each day.9 The relatively low amount in plasma is a result of the high amount of IgA secreted into the gastrointestinal tract. It is estimated that a normal 70-kg adult secretes approximately 2 g of IgA per day.9 IgA also circulates in the plasma as a monomer, dimer, or higher polymer containing approximately 8 percent carbohydrate. Within the IgA class are two major subclasses, designated IgA1 and IgA2. The most abundant subclass is IgA1, which constitutes approximately 85 percent of the total IgA in plasma. The half-life of circulating IgA of either subclass is approximately 6 days.
The primary role for IgA is in mucosal immunity.10 Plasma cells in the lamina propria secrete IgA as a dimer that is held together by a J (joining) chain. The secreted IgA can bind to a poly-Ig receptor, which is an integral membrane glycoprotein expressed on the basal membrane of mucosal cells. Following the binding of IgA, the mucosal epithelial cells mediate endocytosis and transport of the IgA–poly-Ig receptor complex in vesicles that are exported to the epithelial luminal surface. Here the poly-Ig receptor is proteolytically cleaved, releasing the extracellular domain, which remains complexed with the secreted IgA as a 70-kDa secretory protein that can protect the secreted IgA molecule from proteolytic digestion by enzymes in the intestinal lumen. This modified form of IgA, comprised of an IgA dimer bound to the J chain and secretory protein, is the principal antibody in saliva, tears, colostrum, and the fluids of the gastrointestinal, respiratory, and urinary tracts.
IgA can direct various effector functions by cells that bear specific Fc receptors for IgA (FcαR). FcαRI is the principal myeloid IgA receptor and is responsible for directing various IgA-mediated effector responses, such as respiratory burst, degranulation, and phagocytosis by granulocytes, monocytes, or macrophages. Another IgA receptor specific for the secretory protein can elicit powerful effector responses from eosinophils.11 On the other hand, IgA antibodies do not cross the placenta, fix complement via the classic pathway, or bind efficiently to cell surfaces. Their main function may be to prevent foreign substances from adhering to mucosal surfaces and entering the blood.
Defective glycosylation of IgA1 can lead to the most common form glomerulonephritis, namely Berger disease or IgA nephropathy. This is an autoimmune disorder in which neoepitopes caused by defective galactosylation of O-linked glycans in the hinge region of human IgA1 are recognized by antiglycan IgG or IgA1 antibodies.12 Some of the resultant immune complexes in the circulation escape normal clearance mechanisms, deposit in the renal mesangium, and induce glomerular injury.13 Another nephritis associated with glomerular IgA deposits is Henoch-Schönlein purpura, a condition that most commonly presents with a characteristic pruritic skin rash, arthritis, and abdominal pain in children or young adults (Chap. 122).
In a normal adult, approximately 6 percent of the total plasma immunoglobulins belong to the IgM class (see Tables 75–1 and 75–2). IgM molecules classically are termed macroglobulins because of their large molecular weight. Circulating IgM molecules contain 12 percent carbohydrate and are formed through the linkage of five identical immunoglobulin units by disulfide bonds and by a J chain (Fig. 75–2).14 IgM represents the predominant immunoglobulin class formed during a primary immune response. IgM macroglobulins do not penetrate easily into extravascular spaces or readily cross the placenta. Compared to monomeric IgG antibodies, pentavalent IgM antibodies fix complement more efficiently. A single IgM molecule on the surface of a red blood cell can initiate complement-mediated hemolysis. IgM is catabolized rapidly, with a plasma half-life of only 6 days. The monomeric form of IgM, with only two heavy and two light chains, is the major immunoglobulin expressed on the B-cell surface (Fig. 75–3). The IgM monomer represents the ligand-binding part of the receptor. The component that is responsible for signal transduction consists of two glycoproteins, CD79a and CD79b. The cytoplasmic domains of CD79a (Ig-α) and CD79b (Ig-β) contain tyrosine motifs responsible for transduction of signal from the receptor.15
Figure 75–2.
Schematic of an IgM pentamer. IgM has 10 binding sites for antigen, each composed of a heavy-chain variable region (H-chain V region) and a light-chain variable region (L-chain V region). Five bivalent IgM molecules are held together by the single joining (J) chain. Broken red colored lines indicate intrachain and interchain disulfide bonds.
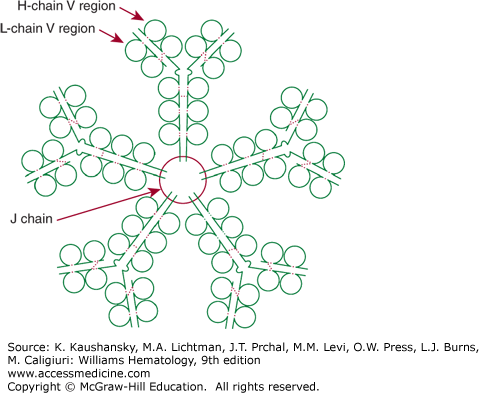
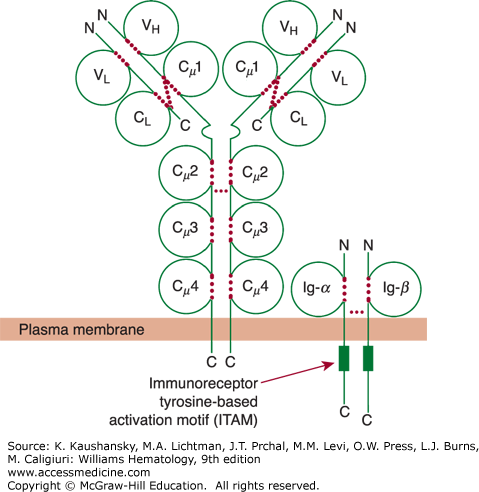
Figure 75–3.
Schematic of membrane IgM and its associated accessory proteins Ig-α (CD79a) and Ig-β (CD79b). The light-chain domains VL and CL and the heavy-chain domains VH, Cμ1 (or CH1), Cμ2 (or CH2), Cμ3 (or CH3), and Cμ4 (or CH4) are labeled inside the respective immunoglobulin domain. Each of the cytoplasmic domains of Ig-α (CD79a) and Ig-β (CD79b) has an immunoreceptor tyrosine-based activation motif (ITAM) depicted by a green rectangle. These ITAMs play a critical role in the signaling events that are initiated by ligation of surface immunoglobulin by antigen. Dotted red colored lines indicate intrachain and interchain disulfide bonds.
IgD is a trace serum protein that composes less than 1 percent of plasma immunoglobulins. IgD is expressed on most peripheral B cells, as is IgM, where it may function as a B-cell membrane receptor for antigen that facilitates recruitment of B cells into specific antigen-driven responses. The molecule has the basic four-chain constant region and contains 11 percent carbohydrate (see Tables 75–1 and 75–2). IgD antibodies are sensitive to proteolytic degradation. They do not penetrate extravascular spaces efficiently, cross the placental barrier, or fix complement via the classic pathway. However, circulating IgD may bind to basophils through a calcium-mobilizing receptor, which, upon crosslinking, can trigger antimicrobial, proinflammatory responses.16 Moreover, crosslinking of basophil-bound IgD also could induce basophils to produce interleukin (IL)-4, IL-13, B-cell activating factor (BAFF, CD272), and a proliferation inducing-ligand (APRIL, CD276), factors that, in turn, could enhance B-cell activation and isotype switching. This might explain why mice made deficient in IgD have fewer B cells, delayed affinity maturation, and weaker production of immunoglobulin isotypes, such as IgE, which are highly dependent on such cytokines.17
Although four human IgE isoforms can be produced by alternative splicing of the epsilon primary transcript,18 each isoform appears to have similar function. IgE has been called reaginic antibody to denote its association with immediate hypersensitivity. It normally constitutes only 0.004 percent of total plasma immunoglobulin (see Tables 75–1 and 75–2). In patients with parasitic infestation and in some children with atopic diseases, plasma IgE levels may rise to 5 to 20 times normal. The IgE molecule consists of a four-chain basic unit plus 12 percent carbohydrate. Monomeric IgE binds via the Fc region to high-affinity receptors on the surface membranes of basophils and mast cells. When bound to tissue mast cells, IgE has a much longer half-life than in plasma, in which its half-life is only approximately 2 days (see Table 75–2). Crosslinking of cell-bound IgE antibody by antigen induces the release of vasoactive amines, lipid-derived inflammatory mediators, proteases, proteoglycans, and cytokines, such as tumor necrosis factor-α (cachectin), interferon-γ, granulocyte-macrophage colony-stimulating factor, and IL-1, IL-3, IL-4, IL-5, and IL-6. These substances act on adjacent cells and may regulate the metabolism of the connective tissue extracellular matrix. These lipid mediators and biogenic amines may produce the rapid components of immediate hypersensitivity, such as vascular leakage, vasodilation, and bronchoconstriction. The released cytokines, on the other hand, are responsible for the late phase of the immediate hypersensitivity response. The physiologic function of this response is not clear. Instead, the immediate hypersensitivity response may represent a pathologic systemic exaggeration of a local physiologic process that ordinarily contributes to the inflammatory response to invading organisms.
Any one of the immunoglobulin isotypes may serve as a B-cell membrane receptor for antigen.15 However, most B cells express surface IgM with or without IgD. Each immunoglobulin is expressed on the surface membrane as a monomer complexed noncovalently with disulfide-linked heterodimeric glycoproteins that, together with surface immunoglobulin, form the B-cell antigen–receptor complex (see Fig. 75–3). For surface IgM, each heterodimer is composed of CD79a, an IgM α-chain of 33 kDa, complexed with CD79b, an Ig β-chain of 37 kDa. CD79a interacts with the transmembrane domain and CH4 domain of the immunoglobulin molecule, which mediates B-cell receptor clustering and signaling in response to antigen (see Fig. 75–3).19 The CD79a chain is a product of the human mb-1 gene (designated CD79a) located at 19q13.2, whereas CD79b is the product of CD79b located on a different chromosome at 17q23. B cells that lack expression of CD79a or CD79b cannot express surface immunoglobulin. CD79a/CD79b are necessary, not only for transport of the assembled immunoglobulin to the cell surface but also for signal transduction following surface immunoglobulin-receptor crosslinking by antigen. Patients with inherited defects in CD79a have an immune deficiency that is indistinguishable from that of classic X-linked agammaglobulinemia (Chap. 80).20 The cytoplasmic tails of CD79a and CD79b each contain immunoreceptor tyrosine-based activation motifs (ITAMs). Such motifs are found in the cytoplasmic domains of several immune system signaling molecules, including those of the T-cell receptor complex (Chap. 76).
B cells can become activated following ligation of their surface immunoglobulin receptors by antigen, which typically is presented on the surface of dendritic cells or macrophages.21,22,23,24 This can cause microclustering of the immunoglobulin receptor complex into the immunologic synapse, which accumulates src family tyrosine kinases (e.g., Lyn, Blk, and Fyn), which can phosphorylate tyrosine residues in the ITAMs of CD79a and CD79b. In turn, the phosphorylated ITAM binds cytoplasmic signaling molecules, the most important of which is p72Syk, a 72-kDa tyrosine kinase. Following its recruitment to the activated immunoglobulin receptor complex, p72Syk itself becomes activated through phosphorylation, allowing it to phosphorylate the cytosolic adapter protein BLNK (B-cell linker protein, also known as SLP-65, BASH, or BCA).25 BLNK serves as a docking site for a number of important signaling molecules, including Bruton tyrosine kinase (BTK), Vav-1, Vav-2, and phospholipase C gamma (PLCγ).26 Dual phosphorylation and activation of PLCγ by BTK and p72syk allows PLCγ to effect hydrolysis of the polyphosphoinositides into inositol-1,4,5-trisphosphate and diacylglycerol, which, in turn, increase intracellular Ca2+ and activate protein kinase C and Ras, respectively. The importance of these activation events in B-cell signaling and development is underscored by patients with inherited defects in BTK, who lack B-cell development and have X-linked agammaglobulinemia (Chap. 80). Furthermore, inhibitors of BTK demonstrate clinical activity in the treatment of patients with various B-cell malignancies, which appear dependent upon constitutive signaling via the immunoglobulin receptor.27
To mitigate the problem of accidental initiation of signal transduction, the signaling cascade is subject to negative controls. The quantity and quality of immunoglobulin receptor signaling are modulated by several transmembrane proteins that are associated with the immunoglobulin–CD79a/CD79b receptor complex.28 These associated proteins can be either costimulatory (e.g., CD19) or inhibitory (e.g., CD22, CD32 [FcγRII], CD72). In contrast to CD79a and CD79b, CD22 and CD72 have cytoplasmic domains with immunoreceptor tyrosine-based inhibitory motifs. When immunoreceptor tyrosine-based inhibitory motifs are phosphorylated by activated Lyn kinase, the domains recruit Src homology 2 (SH2) domain-containing protein tyrosine phosphatase 1 (SHP-1), otherwise known as protein tyrosine phosphatase 1c,29,30 or phosphatidylinositol-3,4,5-triphosphate 5-phosphatase (SHIP-1).31 Bound SHP-1 or SHIP-1 can remove the phosphate group from the phosphorylated (and thereby activated) tyrosine kinases, returning these kinases to their inactive state so that they no longer trigger B-cell activation. The importance of SHP-1 in limiting B cell activation is demonstrated by mutant mice that lack this phosphatase.32 The B lymphocytes of such animals are stimulated by much lower concentrations of antigen than the B lymphocytes of normal mice, causing excessive B-cell proliferation, autoimmune disease, and early mortality.
GENETICS OF IMMUNOGLOBULINS
Immunoglobulin genes are inherited in three unlinked gene complexes: one for the heavy-chain classes, one for κ light chains, and one for λ light chains. The immunoglobulin heavy-chain gene complex is located at band q32 of the long arm of chromosome 14. This complex is composed of 39 functional heavy-chain variable-region (VH) genes, more than 120 nonfunctional VH pseudogenes, 25 functional diversity (D) segments, six functional JH minigenes, and exons encoding the constant regions for each of the immunoglobulin heavy-chain isotypes (Fig. 75–4).33 The κ light-chain gene complex is contained within band p12 on the short arm of chromosome 2. This gene complex consists of approximately 40 functional κ light-chain variable-region genes (Vκ genes), more than 30 nonfunctional Vκ pseudogenes, five Jκ segments, one constant-region exon, and one kappa-deleting element (Kde) (Fig. 75–5). Many of the Vκ genes in the so-called p region most proximal to the Jκ segments are in the opposite orientation of the Jκ segments, thus requiring that the Vκ exons in the proximal region to undergo inversion during immunoglobulin gene rearrangement (Fig. 75–5). The λ light-chain gene complex is located at band q11.2 on the long arm of chromosome 22, 6 Mb from the centromere.34 This gene complex consists of approximately 41 functional λ light-chain variable-region genes (Vλ genes), more than 30 Vλ pseudogenes, four functional λ constant-region genes (Cλ1, Cλ2, Cλ3, Cλ7), and three λ constant-region pseudogenes (Cλ4, Cλ5, Cλ6), each associated with one Jλ segment (Fig. 75–5). The constant-region elements of the heavy-chain gene complex are proximal to variable-region segments on chromosome 14, whereas the constant-region segments of the two light chains are in the opposite orientation, telomeric to the variable-region genes.
Figure 75–4.
Immunoglobulin heavy-chain (IGHC) gene complex. The heavy-chain genes encoding the constant regions are represented by blue boxes. Switch regions are represented by a filled circle upstream of the IGHC genes. Enhancers are represented by light blue circles. Each IGHV, IGHD and IGJH gene is labeled on the right of each symbol. Functional IGHV, IGHD, and IGHJ genes are represented by green boxes or blue and yellow lines, respectively. IGHV, IGJH, and IGHC pseudogenes are represented by red, orange boxes, or blue open boxes, respectively. IGHV and IGHC open-reading frames are represented by yellow and blue dashed boxes, respectively. Unrelated pseudogenes are represented by purple open boxes. Colors are according to IMGT color menu for genes. (Used with permission of Marie-Paule Lefranc. IMGT®, the international ImMunoGeneTics information system® http://www.imgt.org.)

Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


