INTRODUCTION
SUMMARY
Follicular Lymphoma (FL) is an indolent, neoplastic disorder of germinal center-derived B lymphocytes that afflicts approximately 14,000 people in the United States each year. It typically presents as a disseminated disorder with painless, diffuse lymphadenopathy and marrow infiltration, and may be associated with hepatosplenomegaly and circulating lymphoma cells in the blood. A characteristic translocation, t(14;18), is found in the cells of 85 percent of patients, which deregulates BCL2 protein expression and inhibits apoptosis of affected B cells. The cells typically express monoclonal surface immunoglobulin, CD10, CD19, CD20, CD22, CD45, and CD79a on their cell surface, but not CD5 or CD23. Patients are often asymptomatic at the time of presentation, and may live for many years in good health without therapy. On the other hand, most patients eventually develop progressive lymphadenopathy, causing symptoms mandating intervention. Many treatment regimens are effective at inducing remissions, including single-agent rituximab or chlorambucil; or several multidrug programs, including bendamustine plus rituximab (BR), rituximab, cyclophosphamide, vincristine, and prednisone (R-CVP); rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP). None of these therapies, however, is considered curative and most patients eventually relapse with recurrent disease. Autologous and allogeneic hematopoietic cell transplantation (HCT) can induce prolonged remissions in many patients with relapsed FL, but the role of HCT in this disease is controversial. Histologic transformation to aggressive lymphoma occurs in 30 to 40 percent of patients, usually leading to death within a few years of transformation.
Acronyms and Abbreviations
ADCC, antibody-dependent cellular cytotoxicity; BR, bendamustine and rituximab; CDC, complement-dependent cytotoxicity; CHOP, cyclophosphamide, doxorubicin, vincristine, prednisone; CR, complete response; CVP, cyclophosphamide, vincristine, prednisone; FCM, fludarabine, cyclophosphamide, mitoxantrone; FDG, fluoro-2-deoxyglucose; FL, follicular lymphoma; FND, fludarabine, mitoxantrone (Novantrone), dexamethasone; GELF, Groupe d’Etudes des Lymphomes Folliculaires; Gy, gray; HLA, histocompatibility locus antigen; IFN, interferon; IPI, international prognostic index; KLH, keyhole limpet hemocyanin; LDH, lactate dehydrogenase; NHL, non-Hodgkin lymphoma; ORR, overall response rate; OS, overall survival; PCR, polymerase chain reaction; PET, positron emission tomography; PFS, progression-free survival; PR, partial remission; ProMACE/MOPP, prednisone, methotrexate, doxorubicin, cyclophosphamide, etoposide, mechlorethamine, vincristine, procarbazine, prednisone; R-CHOP, rituximab plus CHOP; R-CVP, rituximab plus CVP; RIT, radioimmunotherapy; WHO, World Health Organization.
DEFINITION AND HISTORY
Follicular lymphoma (FL) is an indolent lymphoid neoplasm that is derived from mutated germinal center B cells and exhibits a nodular or follicular histologic pattern. It is typically composed of a mixture of small, cleaved follicle center cells (centrocytes) and large noncleaved follicle center cells (centroblasts). The disease has masqueraded under multiple monikers, including “nodular lymphoma” in the Rappaport classification, and “follicle center cell lymphoma” in the Working Formulation.1 The current World Health Organization (WHO) classification proposes the terms follicular lymphoma, grades 1, 2, and 3, to differentiate cases based on the numbers of centroblasts per high-power microscopic field (see “Lymph Node Morphology and Lymphocyte Immunophenotype” below).2
EPIDEMIOLOGY
FL accounts for approximately 20 to 25 percent of adult non-Hodgkin lymphomas (NHLs) in the United States, with an annual incidence of approximately 14,000 new cases per year.3,4 FL is most common in North America and Western Europe, and much less frequent in Eastern Europe, Asia, Africa, and in Americans of African descent.2 The median age at diagnosis is 59 years, and the male-to-female ratio is 1:1.7. The disease is rare in persons younger than age 20 years, and pediatric cases appear to represent a separate disease entity that is typically localized, lacks the t(14;18) translocation and BCL2 expression, and has a very good prognosis.5,6
CLINICAL FEATURES
Patients with FL usually present with painless diffuse lymphadenopathy. Less frequently, patients may have vague abdominal complaints, including pain, early satiety, and increasing girth, which may be caused by a large abdominal mass or hepatosplenomegaly. Approximately 10 percent of patients present with B symptoms (fever, drenching night sweats, or loss of 10 percent of body weight). The disease usually is widespread at presentation, with involvement of multiple lymph node–bearing sites, liver, and spleen. The marrow is involved in 40 to 70 percent of patients at diagnosis. FL may occasionally present with primary involvement of extranodal sites, such as the skin, gastrointestinal tract, ocular adnexa, and breast, but CNS disease is rare, unless histologic transformation to diffuse large B-cell lymphoma has occurred.2
LABORATORY FEATURES
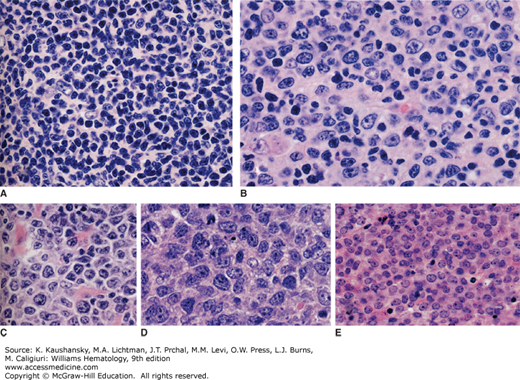
FL exhibits a predominantly nodular lymph node pattern, however, the neoplastic follicles are distorted and as the disease progresses, the malignant follicles efface the nodal architecture (see Chap. 96, Fig. 96–18), commonly resulting in the development of areas of diffuse involvement, which may predominate histologically. The WHO has developed a three-grade system for classifying FL according to the proportion of centroblasts detected microscopically: grade 1 lymphomas have 0 to 5 centroblasts, grade 2 lymphomas have 6 to 15 centroblasts, and grade 3 lymphomas have more than 15 centroblasts per high-power microscopic field (Fig. 99–1).2 Grade 3 FL is further subdivided into grade 3A, in which some small centrocytes are present despite the predominance of centroblasts, and grade 3B, in which solid sheets of centroblasts are exclusively present and centrocytes are entirely absent.2 Some, but not all, studies suggest that grades 1 and 2 lymphomas follow a more indolent course than grade 3 FL, and many authorities suggest that these lower grades should be treated more conservatively than grade 3 FL.7 Other studies indicate a similar natural history for grades 1, 2, and 3A.8 Nearly all authorities now agree, however, that grade 3B FL behaves aggressively and should be treated with anthracycline-containing regimens (e.g., rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone [R-CHOP]) similar to diffuse large B-cell lymphoma.2 FL cells of all grades typically express monoclonal surface immunoglobulin, are positive for BCL-2, BCL6, and CD10, and express the pan–B-cell surface antigens CD19, CD20, CD22, and CD79a, but do not express CD5, CD23, CD11c, or CD43.
Figure 99–1.
Follicular lymphoma grading is based on the relative proportions of small cells (centrocytes) and centroblasts (centroblasts). A. Grade 1 (0–5 centroblasts/high-powered field). B. Grade 2 (6–15 centroblasts/high-powered field). C. Grade 3A (>15 centroblasts/high-powered field). D and E. Grade 3B. See text for further definitions of grades 1, 2, 3A, and 3B. (Reproduced with permission from Harris NL, Swerdlow SH, Jaffe ES, et al: Follicular Lymphoma, in WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, edited by Swerdlow SH, Campo E, Harris NL, et al: p 220–226. International Agency for Research on Cancer, Lyon, 2008.)
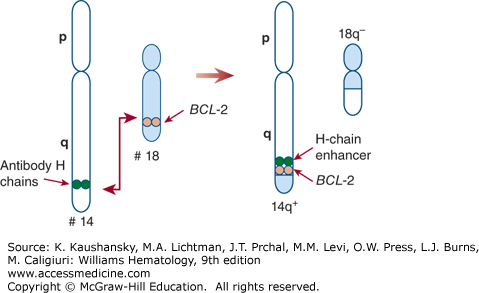
The classic cytogenetic finding detected in FL is the t(14;18)(q32;q21) translocation that juxtaposes the BCL-2 gene on band q21 of chromosome 18 with the immunoglobulin heavy-chain gene on band 32 of chromosome 14 (Fig. 99–2).9 The immunoglobulin enhancer element results in amplified expression of the translocated gene product and, thus, overexpression of BCL-2 protein leading to inhibition of apoptosis of affected B cells. Quantitative real-time polymerase chain reaction (PCR) assays on blood and marrow can determine the number of t(14;18)-expressing cells and may be useful in predicting the outcome of therapy. The t(14;18) translocation is found in approximately 85 percent of patients in the United States, but the translocation is present in a significantly lower percentage of Asian patients afflicted with FL. Detection of the t(14;18) translocation in lymphoid cells is neither necessary nor sufficient for the diagnosis of FL. Small numbers of B cells harboring the t(14;18) translocation can be detected in the blood of 25 to 75 percent of healthy individuals, as well as in reactive lymph nodes and tonsils if a very sensitive nested or reverse-transcription PCR assay is employed.2 Additional cytogenetic abnormalities are found in the cells of 90 percent of patients with FL. The finding of multiple cytogenetic abnormalities is commonly associated with higher histologic grade and with the probability of transformation to aggressive lymphoma. A recent large, high-resolution, genome-wide copy-number analysis demonstrated that common recurrent chromosomal abnormalities include gains of chromosomes 2, 5, 6p, 7, 8, 12, 17q, 18, 21, and X and losses on 6q and 17p.10 Frequent small abnormalities are also commonly observed, including losses of 1p36.33–p36.31,6q23.3–q24.1, and 10q23.1–q25.1 and gains of 2p16.1–p15,8q24.13–q24.3, and 12q12–q13.13. Copy-number abnormalities more commonly observed in transformed FL include gains of 3q27.3–q28 and chromosome 11 and losses of 9p21.3 and 15q.10 Important candidate genes whose expression is affected by these copy-number abnormalities includeTNFRSF14, PRDM16, TP73, and ARIDIA on chromosome 1p36; BCL10 on chromosome 1p; REL and BCL11A on 2p16; BCL6 on chromosome 3q27; histocompatibility locus antigen (HLA)-B, HLA-C, CCND3, and PRDM1 on chromosome 6p21; TNFAIP3 or PERP on chromosome 6q23; CARD11 on chromosome 7p22; MYC on chromosome 8q24; CDKN2A or CDKN2B on chromosome 9p21; STAT6 on 12q13.3; and MDM2 on 12q15.10
Evaluation of FL involves performance of a medical history, physical examination (with attention to the lymph nodes in Waldeyer ring and size and involvement of liver and spleen), laboratory testing (including a complete blood count, examination of the blood film and a differential white cell count, lactic dehydrogenase [LDH], β2-microglobulin, comprehensive metabolic panel, serum uric acid level); lymph node biopsy; marrow aspiration and biopsy; flow cytometric analysis of blood, marrow, and lymph node cells; and computed tomography (CT) of the chest, abdomen, and pelvis.7 Excisional lymph node biopsies are strongly preferred for the initial histologic diagnosis of FL, although in cases in which nodal masses are inaccessible, generous needle-core biopsies may suffice. The diagnosis should not be established merely on the basis of flow cytometry of the blood or marrow, or on cytologic examination of aspiration needle biopsies of lymph node or other tissue.11 Hepatitis B serology should be assessed if rituximab therapy is contemplated, as hepatitis reactivation with rituximab may occasionally be life-threatening. In selected circumstances, additional CT scans of the neck, measurement of the cardiac ejection fraction, serum protein electrophoresis, quantitative immunoglobulins, and hepatitis C testing may be useful. Patients for whom chemotherapy is contemplated should receive counseling regarding contraception, fertility issues, and sperm or egg banking. The role of fluoro-2-deoxyglucose (FDG)-positron emission tomography (PET)/CT imaging in FL is rapidly evolving. Although PET/CT imaging was previously considered optional in FL, recent studies suggest that PET-negativity at the completion of induction chemotherapy is one of the most powerful predictors of both progression-free survival (PFS) and overall survival (OS) in this disease.12
PROGNOSTIC FACTORS
An international working group developed an international prognostic index (IPI) based on five independent variables (age, stage, LDH level, performance status, and number of extranodal sites) that affected OS of aggressive lymphoma patients treated with anthracycline-based combination chemotherapy.13 The IPI was subsequently applied retrospectively to FL and found to be predictive of both OS and PFS for FL (as well as diffuse large B-cell lymphoma). Nevertheless, the IPI was considered to be suboptimal for segregating indolent lymphoma patients into prognostic categories because only 10 to 15 percent of patients with FL fall into the poor risk category using this index. To redress this deficiency, a French cooperative group conducted a detailed prognostic factor analysis of 4167 patients with FL diagnosed between 1985 and 1992 for whom prolonged followup was available to assess OS.14 Five adverse prognostic factors were detected: age (>60 years vs. ≤60 years), Ann Arbor stage (III to IV vs. I to II), hemoglobin level (<120 g/L vs. ≥120 g/L), number of nodal areas (>4 vs. ≤4), and serum LDH level (high vs. normal). Three risk groups were defined: low risk (zero to one adverse factors, 36% of patients), intermediate risk (two adverse factors, 37% of patients, hazard ratio [HR] = 2.3), and poor risk (three or more adverse factors, 27% of patients, HR = 4.3). The Follicular Lymphoma International Prognostic Index (FLIPI) discriminated outcomes for FL better than the IPI, both in the original cohort and in later studies evaluating patients treated with modern combined rituximab-chemotherapy regimens (Fig. 99–3).15 A revised version of the FLIPI index (FLIPI2) was subsequently proposed to address perceived deficiencies in the original model.16 The FLIPI2 model is also based on assessment of five adverse risk factors, namely the presence or absence of an elevated β2-microglobulin level, the longest diameter of the largest involved lymph node (>6 cm), presence of marrow involvement, hemoglobin level less than 12 g/dL, and age older than 60 years. Although several studies have demonstrated the superiority of the FLIPI2 model compared to the original FLIPI model, it has not been widely adopted in North America. A simpler prognostic model was developed based solely on the baseline serum LDH and β2-microglobulin level that has been shown to be superior to the original FLIPI model and equivalent to the FLIPI2 model in prognostic power for predicting outcomes for FL patients.17,18
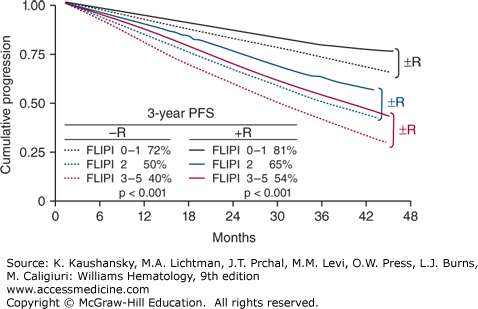
Figure 99–3.
Progression-free survival (PFS) of 827 patients with FL stratified by the Follicular Lymphoma International Prognostic Index (FLIPI) into low risk (0 to 1 risk factors, 40% of patients, black lines), intermediate risk (2 risk factors, 33% of patients, blue lines), or high risk (3 to 5 risk factors, 27% of patients, red lines). Of the 827 patients, 267 were treated with chemotherapy regimens without rituximab (dotted lines) and 560 were treated with rituximab-containing regimens (solid lines). (Data from Federico M, Bellei M, Pro B: Revalidation of FLIPI in patients with follicular lymphoma registered in the F2 study and treated upfront with immunochemotherapy. Proc Am Soc Clin Oncol 25:443s, 2007.)
New molecular approaches are revolutionizing our understanding of the pathogenesis of FL and providing insights into the pathways that might be targeted in the future by rationally designed therapies. Early gene-expression profiling studies of biopsy specimens from patients with untreated FL identified two gene-expression signatures that allowed construction of a survival predictor enabling segregation of patients into four quartiles with disparate median lengths of survival (13.6, 11.1, 10.8, and 3.9 years), independent of clinical prognostic variables.19 One signature (“immune response 1”) was associated with a good prognosis and included genes encoding T-cell markers (e.g., CD7, CD8B1, ITK, LEF1, and STAT4) as well as genes that are highly expressed in macrophages (e.g., ACTN1 and TNFSF13B). The “immune response 2” signature was associated with a poor prognosis and included genes known to be preferentially expressed in macrophages, dendritic cells, or both (e.g., TLR5, FCGR1A, SEPT10, LGMN, and C3AR1). Flow cytometry and cell sorting confirmed that these signatures reflected gene expression by nonmalignant tumor-infiltrating immune cells (CD19-negative cells) and not by the FL cells themselves (CD19-positive cells). The length of survival correlated with the molecular features of the nonmalignant immune cells present in the tumor at diagnosis and presumably reflected the robustness of the immune response mounted against the tumor.
“Next-generation sequencing” studies employing whole-genome or whole-exome sequencing and targeted mutational analyses have demonstrated that somatic mutations in epigenetic regulators are present in almost all cases of FL, including MLL2 (also known as KMT2D), a histone methyltransferase that is mutated in 89 percent of FL; CREBBP and EP300, two highly related histone and nonhistone acetyltransferases that act as transcriptional coactivators in multiple signaling pathways that are mutated in 30 percent and 11 percent of FL, respectively; EZH2, the catalytic subunit of PRC2 that catalyzes trimethylation of lysine 27 on histone H3, a repressive chromatin mark that is mutated in 27 percent of FL; and MEF2B, a calcium-regulated gene that cooperates with CREBBP and EP300 in acetylating histones that is mutated in 15 percent of FL.20,21,22,23,24 Other mutational targets include genes involved in immune modulation (β2-microglobulin, CD58, and TNFRSF14), Janus kinase (JAK)-signal transducer and activator of transcription (STAT) signaling (SOCS1 and STAT6) and B-cell receptor–nuclear factor (NF)-κB signaling (BCL10, CARD11 and CD79B).22 Mutations in CREBBP and EP300 lesions are usually heterozygous, suggesting a haploinsufficient role in tumor suppression, apparently by impairing acetylation-mediated inactivation of the BCL6 oncoprotein and activation of the p53 tumor suppressor.21 A unifying model of the genetic evolution of FL postulates that the t(14;18) translocation serves as a “founder mutation” that is succeeded by development of additional “driver mutations” (e.g., CREBBP), irrelevant “passenger” mutations, and accelerator mutations (TNFRSF14).22,23 Disagreement exists over whether MLL2/KMT2D mutations are “driver” or “accelerator” mutations.22,23 Mutations in EBF1 and in regulators of NF-κB signaling (e.g., MYD88, TNFAIP3) appear to be acquired at the time of histologic transformation to diffuse large B-cell lymphoma.22
THERAPY
Patients with stage I or II FL represent only 10 to 30 percent of cases in most series.2,4 Standard management for stage I or limited contiguous stage II FL involves the administration of involved field radiotherapy (35 to 40 Gray [Gy]).7 Adjuvant chemotherapy does not appear to improve survival in this setting, although some studies suggest that combined chemoradiotherapy may improve PFS. A retrospective review of 177 patients with stage I or II and grade 1 or 2 FL reported a median survival of 14 years following radiation therapy as a single modality.27 Approximately 50 percent of the patients were relapse-free after 5 to 10 years.
Excellent survival has also been observed in highly selected patients with early stage FL who received no initial therapy.25 In a group of 43 patients, 56 percent were free from the requirement for treatment for at least 10 years and 86 percent were alive 10 years after diagnosis. Based on this study, many authorities have concluded that “watchful waiting” is an acceptable alternative to radiotherapy for stage I or II FL. A watchful waiting approach may be particularly appropriate for certain variants of FL, such as FL presenting in the small intestine, which pursues a remarkably indolent course, rarely exhibits progressive growth, very rarely disseminates (two of 63 patients) and does not transform to high-grade disease.26
A large observational study, the National LymphoCare Study, assessed the outcomes of 471 patients with stage I FL according to treatment administered, including rituximab plus chemotherapy (28 percent of patients), radiotherapy alone (27 percent), observation (17 percent), systemic therapy + radiotherapy (13 percent), rituximab monotherapy (12 percent), and other treatments (3 percent).28 This large, prospectively enrolled group of patients showed that national guidelines endorsing radiotherapy alone for stage I FL were not followed by practicing clinicians in the majority of cases. All treatment approaches resulted in excellent outcomes, though PFS was significantly better after a median followup of 57 months in patients treated with either rituximab plus chemotherapy (84 percent) or systemic therapy plus radiotherapy (96 percent) than in patients receiving radiotherapy alone (68 percent).28 This study challenges the paradigm that radiotherapy alone should be the standard of care for patients with early stage indolent lymphoma, although the observational nature of this study, without randomization to treatment arm and the absence of differences in OS, attenuates the impact of the study.
Many patients with FL, particularly grades 1 or 2, will exhibit an indolent, asymptomatic course despite the absence of therapy. Because there is no conclusive evidence that survival of FL patients is improved by immediate institution of therapy, or that conventional management (other than allogeneic stem cell transplantation) can cure the disease, a “watch-and-wait” approach is often recommended for patients with extensive stage II or stage III or IV FL. In one study, survival was 82 percent at 5 years and 73 percent at 10 years after an initial strategy of observation alone, and the median time until therapy was required was 3 years.29 Spontaneous regressions occurred in 23 percent of untreated patients. No differences in survival were observed in a trial of 309 patients randomized to initial watchful waiting or to chlorambucil.30 In another trial, patients were randomized to either watchful waiting or to immediate aggressive combination chemotherapy with prednisone, methotrexate, doxorubicin, cyclophosphamide, etoposide, mechlorethamine, vincristine, procarbazine, prednisone (ProMACE/MOPP) chemotherapy followed by total nodal irradiation.31 The OS rates for the two groups were similar, although the disease-free survival rate was naturally higher in the patients treated with combined modality therapy. Criteria established by the Groupe d’Etudes des Lymphomes Folliculaires (GELF) are useful to identify patients who may benefit from intervention rather than “watchful waiting.” These criteria suggest that treatment is likely to be required for patients with a maximum diameter of any site of disease greater than 7 cm, more than three nodal sites greater than 3 cm in diameter, systemic B symptoms, a spleen size greater than 16 cm, pleural effusions, local compressive symptoms, circulating lymphoma cells, or cytopenias as a result of the lymphoma.32
FL patients can be palliated effectively with a variety of single chemotherapy agents (Table 99–1). Responses to single-agent therapy, such as chlorambucil, a nucleoside analogue, or bendamustine, range from 70 to 90 percent and may last for several years.30,33
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


