Folate antagonists
Peter D. Cole, MD  Lisa Figueiredo, MD
Lisa Figueiredo, MD  Joseph R. Bertino, MD
Joseph R. Bertino, MD
Overview
Folic acid antagonists (antifols) are cytotoxic drugs used as antineoplastic, antimicrobial, anti-inflammatory, and immune-suppressive agents. While several folate antagonists have been developed, methotrexate (4-amino-4-deoxy-10-N-methyl-pteroylglutamic acid; MTX) is the antifol with the most extensive history and widest spectrum of use. MTX remains an essential drug in curative chemotherapy regimens used to treat patients with acute lymphoblastic leukemia, osteosarcoma, and choriocarcinoma and is an important agent in the therapy of patients with lymphoma, breast cancer, bladder cancer, and head and neck cancer. In addition, it is used for patients with nonmalignant diseases such as rheumatoid arthritis, psoriasis, autoimmune diseases, and graft versus host disease. This chapter will review the clinical use of and the metabolism of MTX and discuss structurally related folate antagonists that have been developed to overcome resistance or have alternate intracellular targets.
Historical overview
In the early 1940s, the combined observations that patients with acute leukemia often have serum folate deficiency and that the bone marrow megaloblasts of folate-deficient patients morphologically resemble leukemic blasts prompted some investigators to postulate that leukemia might be a result of a deficiency of this B vitamin. However, it rapidly became apparent that administration of folic acid to patients with leukemia was not only ineffective but often accelerated the course of the disease.1 Thus, efforts to treat these leukemias turned to pharmacologically mimicking folate deficiency using folate analogs with effects antagonistic to those of the vitamin. Aminopterin (4-amino-4-deoxy PGA; AMT; Figure 1) was the first of these analogs to produce temporary remissions in 5 of 16 patients with acute leukemia.2 This report was a landmark in cancer chemotherapy, as the first successful example of the power of rational drug design leading to an effective antineoplastic agent.
Since the initial study indicating the usefulness of AMT in the treatment of acute leukemia of childhood, there has been sustained interest in folate antagonists. Although known to be less potent, 4-amino-4-deoxy-10-N-methyl-pteroylglutamic acid (MTX) supplanted AMT in the clinic in the early 1950s because the toxicity caused by AMT was greater and less predictable.3–5 Newer antifols, rationally designed analogs of folate or MTX, have been synthesized either in an effort to overcome cellular resistance to MTX or to target alternative folate-dependent processes. This chapter will discuss two, pralatrexate and pemetrexed, which recently received FDA approval for oncologic indications.
Mechanisms of action of MTX
Folate antagonists function in several ways: by competing with folates for uptake into cells, by inhibiting the formation of folate coenzymes, or by inhibiting one or more reactions that are mediated by folate coenzymes. Thus far, the clinically important anti-neoplastic folate analogs appear to work primarily by inhibiting dihydrofolate reductase (DHFR) or thymidylate synthase (TS). The prototypic DHFR inhibitor is a 4-amino-substituted pterin compound, such as MTX or AMT (Figure 1). Substitution of an amino group for the 4-hydroxy moiety results in a folate analog with a several thousand-fold increase in affinity for DHFR. The Ki of MTX for DHFR is below 10−10 M, well below the micromolar Km of the natural substrate, dihydrofolate. By stoichiometrically inhibiting DHFR at slightly acidic pH, MTX blocks the cell’s ability to replenish a supply of reduced folates necessary for de novo thymidylate synthesis (Figure 2).6 In rapidly dividing cells, the inhibition of thymidylate biosynthesis leads to a decrease in thymidine triphosphate pools, a decrease in DNA synthesis, and eventually cell death.7
Intracellular metabolism of classical antifols such as MTX to polyglutamate species significantly impacts their function and mechanisms of cytotoxicity.8 Folylpolyglutamate synthetase (FPGS) adds glutamate residues in γ-carboxyl linkage to both folate coenzymes and classical folate antagonists (those with a glutamate moiety). This addition of up to seven or eight additional glutamate molecules serves to add mass and negative charge, markedly reducing efflux and increasing total intracellular accumulation at steady state.9 Both quantitative differences in FPGS expression and qualitative differences in FPGS function10 exist between neoplastic and non-neoplastic tissues, which may explain some of the selectivity of antifolates for neoplastic cells.11 A relative lack of FPGS may explain the observation that a cell population with a large number of G0 cells would be less affected by the same concentration and time of exposure to MTX than a population with more actively dividing cells.
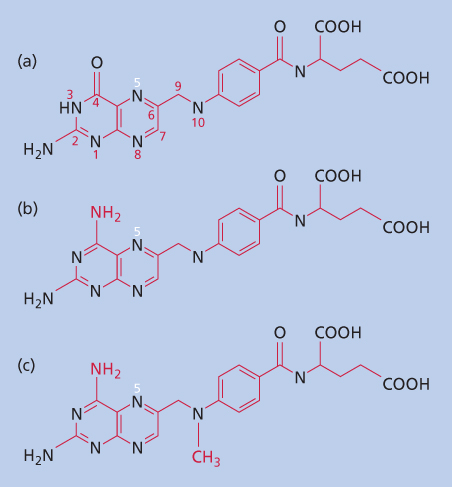
Figure 1 Structure of folic acid and structurally related classical antifols, AMT and MTX. (a) Folic acid (pteroylglutamic acid; PGA). (b) Aminopterin (4-amino-PGA). (c) Methotrexate (4-amino-N-10-methyl PGA).
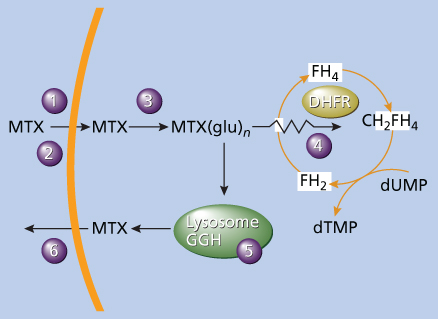
Figure 2 Primary site of action of MTX and MTX(glu)n. MTX enters cells by either the reduced folate carrier (1) or the membrane folate-binding protein (2). MTX is then metabolized by the cytosolic enzyme folylpolyglutamate synthetase (3) to MTX(glu)n, a potent inhibitor of dihydrofolate reductase (DHFR) (4). MTX(glu)n can be hydrolyzed to MTX by the lysosomal enzyme γ-glutamyl hydrolase (GGH) (5). Abbreviations: CH2FH4, N5, N10-methylene tetrahydrofolate; dTMP, deoxythymidine monophosphate; dUMP, deoxyuridine monophosphate/deoxyuridylate; FH2, dihydrofolate; FH4, tetrahydrofolate; MTX, methotrexate; MTX(glu)n, MTX polyglutamates.
MTX polyglutamates are more potent inhibitors of DHFR than the parent compound because they bind as tightly to DHFR as MTX but dissociate less rapidly.12 In addition, MTX polyglutamates are potent inhibitors of other folate-requiring enzymes, including TS13 and two of the rate-limiting steps of de novo purine synthesis: glycinamide ribonucleotide (GAR) and aminoimidazole carboxamide ribonucleotide (AICAR) transformylases.14 These two enzymes are potently inhibited by DHF-polyglutamates and 10-formyl-DHF polyglutamates, which increase after MTX inhibits DHFR.15 As a result, inhibition of de novo purine synthesis may be at least as relevant as DHFR inhibition to the cytotoxic effects of MTX in cancer cells16 and for the anti-inflammatory action of MTX in patients with rheumatologic diseases.17, 18
Other possible mechanisms by which MTX exerts antineoplastic or anti-inflammatory action are worth mentioning. First, by inhibiting folate-dependent methionine biosynthesis, MTX causes intracellular concentrations of homocysteine (Hcy) to increase, resulting in a secondary increase in S-adenosyl-homocysteine (SAH), a potent inhibitor of many folate-dependent methylation reactions. MTX exposure, therefore, can block membrane localization of ras,19 a member of a family of critical signal transduction proteins constitutively activated in a number of human cancers. Second, the anti-inflammatory effects of MTX and some component of its antineoplastic activity may be due to its ability to inhibit endothelial cell proliferation at low concentrations.20 Preclinical data confirm that low-dose methotrexate can inhibit the growth of microscopic metastatic disease through its antiangiogenic properties.21 In addition to the mechanisms mentioned earlier, rapidly proliferating cancer cells overexpress enzymes of the mitochondrial and glycine-serine pathway, and this phenotype is a recently appreciated determinant of methotrexate sensitivity.22
Pharmacokinetics of MTX
MTX is one of few anticancer agents for which pharmacokinetic data are routinely used in clinical practice to modulate the balance between efficacy and toxicity.23 Retrospective analysis of children with acute lymphoblastic leukemia (ALL) shows that lower MTX clearance24 and higher MTX concentrations25 are associated with lower risk of relapse. Even more intriguing are data from a prospective randomized trial in patients with ALL comparing dosing by body surface area with individualized dosing based on pharmacokinetic data, which showed significantly improved complete continuous remission rates in the individualized therapy arm.26 It is possible, however, that these results are protocol specific, as others have found that pharmacologically guided treatment intensification led to inferior outcomes for some subpopulations.27
Absorption
Following oral administration, peak plasma concentrations occur 1–5 h after a dose of 15–30 mg/m2. Absorption can be relatively poor and unpredictable,28, 29 affected by food, nonabsorbable antibiotics, bile salts, and a shortened intestine transit time. Thus, it is suggested that the drug be taken on an empty stomach with clear liquids. Nevertheless, at a dose and schedule of 25 mg/m2, given orally every 6 h for four doses, plasma MTX concentration >0.5 μM was seen in more than 85% of pediatric patients with ALL, indicating the reliability of this oral regimen.30
Distribution
After intravenous (IV) administration of MTX, the initial volume of distribution (Vd) is approximately 0.18 L/kg of body weight. The initial distribution phase has a t1/2 of 30–45 min; the beta t1/2 is 3–4 h. Steady-state Vd is between 0.4 and 0.8 L/kg.31
After high doses of MTX (>3 g/m2), peak serum concentrations in the range 10−4 to 10−3 M are achieved.32 At these concentrations, transmembrane transport is saturated, limiting further influx of MTX to passive diffusion. Uptake of reduced folates, including leucovorin (LV), is inhibited as well. Studies of MTX metabolism in lymphoblasts in vitro have also shown that too high an extracellular concentration of drug can impede metabolism of MTX to a polyglutamate.33
MTX binding to plasma proteins, especially to albumin, is approximately 50%.34, 35 The 7-hydroxy metabolite of MTX is 90% bound to plasma proteins, but apparently does not interfere with MTX binding to plasma proteins at clinically observed concentrations. The highest tissue-to-plasma concentrations found in humans are in the liver and kidney, followed by the gastrointestinal tract. Prolonged plasma levels after high-dose MTX infusions in humans have been attributed to decreased transit rate secondary to gastrointestinal obstruction.
Because of the blood–brain barrier and efflux mechanisms that actively remove MTX from the CNS,36 cerebrospinal fluid (CSF) MTX concentrations are approximately 1% of those in the plasma; therefore, cytocidal concentrations are not obtained in the CSF after conventional doses but only with doses of 500 mg/m2 and higher.37 After high-dose systemic MTX administration, lumbar CSF and ventricular CSF concentrations were similar. HDMTX may be able to replace intrathecal drug for the treatment of patients with nonleukemic leptomeningeal disease.38 However, a recent meta-analysis of CNS-directed therapy for children with ALL concluded that efforts to increase CSF penetration using HDMTX have not produced the desired result of lowering the rate of CNS relapse in this population.39
As MTX is accumulated poorly into the CSF, even small doses of LV given orally can increase CSF folates significantly. This systemic rescue, especially if given too early after MTX, may rescue leukemic cells in the CSF compartment.40
When injected into an indwelling ventricular catheter, MTX reaches reproducible therapeutic drug concentrations (>10−6 M) for at least 48 h.41 In contrast, when MTX is given by the lumbar route into the CSF, it distributes unreliably into the ventricles. An improved dose schedule utilizing the administration of multiple small doses of intrathecal MTX has been suggested.42 Following intrathecal administration, MTX slowly exits into the systemic circulation with a t1/2 of 8–10 h.36 Systemic toxicity can be observed if multiple doses of intrathecal MTX are administered without LV rescue. The pharmacology of intrathecal MTX and the amount of intraventricular MTX may be altered by overt meningeal leukemia and the position of the patient at the time of lumbar puncture.43 The clinical observation that irradiation followed by MTX treatment may predispose patients to neurotoxicity may be a consequence of the effect of radiation therapy on the blood–brain barrier.44
Patients with pleural or peritoneal effusions may be at increased risk for developing toxicity to HD-MTX as a result of “third spacing” or MTX trapping in the infusion and slow release leading to sustained MTX concentrations in serum.45 In these circumstances, higher LV doses and prolonged LV rescue may be necessary, until the serum level of MTX decreases to <0.05 × 10−6 M.
Metabolism
The major metabolite of MTX, produced by the action of hepatic aldehyde oxidase, is 7-hydroxy MTX (7-OH MTX) (Figure 3), which is only 1% as potent an inhibitor of DHFR as MTX.46 It is also less water soluble than MTX and may contribute to renal toxicity after high doses.47
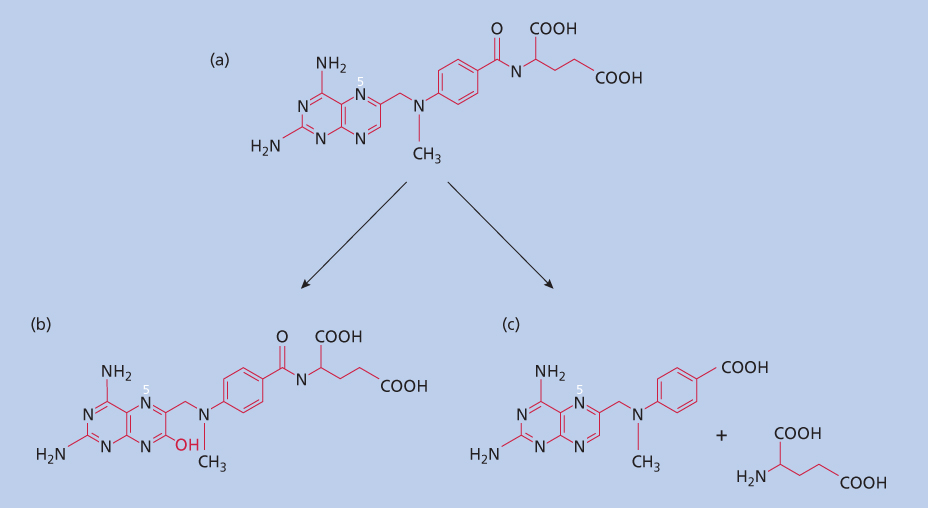
Figure 3 Catabolism of MTX. MTX (a) can be converted in the liver to 7-OH MTX (b). In addition, enteric bacteria will cleave the molecule to dAMPA plus glutamate (c).
A second, less important pathway of metabolism of MTX occurs in the intestine. MTX is hydrolyzed by bacteria to the pteroate (4-deoxy-4-amino-N10-methyl pteroic acid; dAMPA) and glutamic acid (Figure 3).48 dAMPA, like 7-OH MTX, is also a relatively inactive metabolite with approximately 1/200th the affinity of MTX for DHFR. dAMPA excretion in the urine accounts for <5% of the dose administered.
The third metabolic product of MTX is MTX polyglutamate. As discussed earlier, MTX polyglutamates are at least as potent inhibitors of DHFR as is MTX and have a slower rate of disassociation from DHFR.12 MTX polyglutamates are not found in plasma or urine because of the abundant activity of γ-glutamyl hydrolase(s) (GGH) in plasma that convert folyl- and MTX-polyglutamates to monoglutamates. Similar to MTX, 7-OH MTX is also polyglutamylated intracellularly, and retention of these polyglutamate forms could contribute to MTX cytotoxicity.49
Compliance with oral MTX regimens can be monitored by measuring MTX-polyglutamate concentrations within circulating erythrocytes red blood cells (RBCs).50–52 Nucleated RBC precursors within the bone marrow will accumulate and metabolize circulating MTX. The resulting MTX polyglutamates will remain within the mature RBC throughout its lifespan,53 while unmetabolized MTX will gradually efflux.54
Excretion
The majority of administered MTX (and its metabolites 7-OH MTX and DAMPA) is excreted unchanged in the urine.55, 56 Because of active secretion in the proximal tubules, renal clearance of MTX can exceed creatinine clearance.57 There is wide inter-patient variability in MTX clearance, which does not correlate perfectly with renal function.58 MTX excretion is reported to be inversely related to age as plasma clearance of renal eliminated drugs is closely correlated with maturation of kidney function. Children tend to exhibit more rapid MTX clearance than adults, and a general trend toward decreasing clearance with increasing age has been observed.59 MTX excretion through organic acid transporters can be inhibited by probenecid or competitively blocked by other weak organic acids, such as aspirin or penicillin G. MTX elimination is increased by drugs that block distal tubular reabsorption, such as folic acid, some cephalosporins, and sulfamethoxazole.
Less than 10% of MTX is typically recovered in the feces.60 Following IV administration of doses of 30–80 mg/m2, 0.4–20% of the administered dose is excreted through the canalicular multiorganic acid transporter (cMOAT; ABCC2; MRP2) into the bile. Using a knock-out mouse model (ABCC2−/−; ABCG2−/−), it was shown that both transporters play overlapping roles in the elimination of MTX and 7-OH-MTX. Many polymorphisms and mutations of these transporters have been identified, leading to differences in expression and activity, affecting systemic MTX exposure. Consequently, patients with mutations or heterozygous polymorphisms in ABCC2 and/or ABCG2 might be at increased risk for MTX toxicity.61
Drug interactions
Several drugs used in cancer patients, including antibiotics, may alter the renal excretion of MTX, increasing toxicity or decreasing efficacy. Deleterious and even fatal reactions have been reported between MTX and nonsteroidal anti-inflammatory drugs, in particular with naproxen and ketoprofen.62, 63 This increased toxicity may be due to decreased renal elimination, possibly as a result of competition for renal secretion.57 Other commonly used organic drugs may also potentiate MTX toxicity, such as phenylbutazone, salicylate, and probenecid.64, 65 Probenecid increased the efficacy of MTX in tumor-bearing mice, but it has not been used clinically with this goal in mind.66
Increased toxicity was also reported when trimethoprim, the antibacterial agent, was used together with MTX. Presumably, this antifolate, with only weak binding affinity to mammalian DHFR, lowers folate stores, especially in patients with subclinical folate deficiency, making marrow cells more susceptible to MTX-induced toxicity.67 Alcohol should also be avoided in patients receiving MTX because of the risk of hepatic fibrosis and cirrhosis.
Pharmacogenomics
A growing body of data implicates inherited variation in genes for enzymes responsible for folate metabolism in interpatient variability in antifolate response or toxicity. A more detailed discussion of these data is beyond the scope of this chapter but has been the subject of comprehensive reviews.68–71 Briefly, functional polymorphisms have been described in either the promoter or coding regions of the genes for DHFR, methylenetetrahydrofolate reductase (MTHFR), aminoimidazole carboxamide ribonucleotide transformylase (ATIC), the reduced folate carrier (RFC), GGH, methionine synthase (MTR), methionine synthase reductase (MTRR), methylenetetrahydrofolate dehydrogenase (MTHFD), serine hydroxymethyltransferase (SHMT), TS, and solute carrier organic anion transporter gene 1B1 (SLCO1B1). Many of these polymorphisms are present at significant frequency among the population and some have been linked to higher rates of relapse or toxicity among patients with acute lymphoblastic leukemia72–77 or rheumatoid arthritis.78 If replicated in larger populations, these data suggest the potential for individualizing MTX therapy, based on each patient’s genotype.
Gene–environment interactions may also modulate the effects of genotypic variation on toxicity. To focus on one relevant example, some of the observed variation in serum homocysteine (a marker of functional folate deficiency) is explained by two common functional polymorphisms in the MTHFR gene, C677T and A1298C, but only under conditions of decreased intake of dietary folate.79, 80 Adequate dietary folate in countries with mandated folate supplementation could erase the effects of genetic polymorphisms.
Clinical application
Clinical dosage schedules
MTX has been administered on a variety of dosage schedules since its introduction into the clinic more than six decades ago (Table 1). In a trial of MTX in patients with head and neck cancer treated with 50, 500, or 5000 mg/m2 with LV “rescue,” a trend of dose responsiveness was seen (5 of 24, 5 of 16, 9 of 18, respectively). Some responses were noted with the 5000 mg/m2 dose regimen in patients who did not respond at lower doses.81 The importance of dose scheduling was emphasized by an experimental study showing that resistance to high-dose pulse MTX may not extend to continuous low-dose exposure.82 Determining the optimum dose schedule of MTX is complicated by the use of the drug in combination therapy (Table 2). Sequencing appears to be important when MTX is used with 5-fluorouracil (5-FU), with L-asparaginase, and probably with cytosine arabinoside and 6-mercaptopurine or 6-thioguanine. Table 2 summarizes the use of some common drug combinations that include MTX, along with sequence specificity.
Table 1 Dosage schedules commonly used for methotrexate (MTX)
| Schedule and dose | Use/comments |
| Oral | |
| Weekly or biweekly (15–25 mg in single or divided doses) | Mainly for nonmalignant conditions, such as psoriasis or rheumatoid arthritis |
| Weekly or biweekly (20–30 mg/m2) | Maintenance therapy for ALL |
| Parenteral | |
| Pulse weekly (30–60 mg/m2) | Choriocarcinoma, ALL |
| Intermediate dose (120–500 mg/m2 weekly) | ALL, NHL; requires LV rescue, 10–15 mg/m2 q 6 h × 6–8 doses |
| High dose (500–12,000 mg/m2 weekly or every other week) | Osteosarcoma, ALL, neoplastic meningitis; requires LV rescue |
Abbreviations: ALL, acute lymphoblastic leukemia; LV, leucovorin; NHL, non-Hodgkin lymphoma.
Table 2 Combination chemotherapy with methotrexate (MTX)
| Used with | Schedule notes | Result | Comments |
| 5-FU | MTX must precede 5-FU by 24 h | Synergistic | |
| Anthracyclines | Additive | ||
| Bleomycin | Additive | Mucosal toxicity is increased | |
| Corticosteroids | Used together | Synergistic | Used in ALL |
| Cyclophosphamide | Used together | Additive | |
| Cytarabine | Used together | Additive or synergistic | |
| L-asparaginase | If MTX precedes l-asparaginase by 24 h If used simultaneously | Synergistic Antagonistic | Used in ALL, AML |
| Vinca alkaloids | Additive |
Abbreviations: ALL, acute lymphoblastic leukemia; AML, acute myelogenous leukemia; 5-FU, 5-fl uorouracil.
Current uses for MTX in the treatment of neoplastic disease
Acute lymphoblastic leukemia
MTX is a component of nearly all multiagent therapeutic regimens for patients with acute lymphoblastic leukemia post-remission, and some protocols include MTX in remission induction. In addition to systemic use, MTX is administered intrathecally for the treatment of meningeal leukemia and for prophylaxis against CNS relapse.
During the intensive, early post-remission phases, MTX can be administered orally or parenterally. Parenteral administration at intermediate dose (100–500 mg/m2/dose) or high dose (≥1000 mg/m2) has been incorporated in some protocols to increase accumulation of MTX-polyglutamates by blast cells,83 to overcome mechanisms of resistance and to increase penetration into protected sites including the CNS and testes.84 While the ability of HD-MTX to prevent CNS relapse is not clearly proven,39 the rate of isolated testicular relapse does appear to have decreased with the addition of intermediate or HD-MTX.85 Randomized trials comparing escalating IV doses with oral MTX showed a significant improvement in 5-year event-free survival (EFS) in those patients randomly assigned to the IV MTX-based interim maintenance compared with the oral MTX-based arm.86–89 However, in some studies, the increase in EFS came at the expense of increased hematologic and neurologic toxicity.90–93
During later maintenance phases, most current protocols rely on prolonged weekly administration of MTX at low doses (20–50 mg/m2/dose) in combination with daily mercaptopurine. Early studies showed that twice-weekly therapy (20 mg/m2) was superior to continuous daily oral administration for treatment during remission.94 The effectiveness of an oral divided dose (25–30 mg/m2 given every 6 h for 4–6 doses weekly) has also been shown.30
Acute myelogenous leukemia
MTX has limited value in the current treatment of patients with acute nonlymphocytic leukemia. Complete remissions can be seen in approximately one quarter of adults with AML treated with MTX-containing regimens: prednisone, Oncovin, methotrexate, and Purinethol (POMP)95 or MTX with L-asparaginase (the “Capizzi Regimen”).96, 97 High-dose regimens with LV rescue have a transient but rapid effect on the peripheral blood count without producing marrow remissions in the large majority of these patients.98 The lack of efficacy of MTX in this disease has been attributed to poor intracellular retention of the drug caused by a lack of polyglutamylation and an increase of the target enzyme DHFR following treatment.99
Lymphoma
On the basis of the phase II studies that indicated that moderate to high doses of MTX (200 mg/m2 to 3 gm/m2) with LV rescue could produce transient regressions in patients with large cell lymphoma, MTX with LV rescue has been added to combination regimens for intermediate- and high-grade lymphomas. In some regimens (e.g., M-BACOD), MTX is used with LV during the leukopenic phase of drug treatment, as the MTX/LV combination has little marrow toxicity.100 On the basis of the experimental studies showing that MTX and cytosine arabinoside produce additive and possibly synergistic effects,101 this combination has also been utilized in regimens to treat this disease (e.g., COMLA; cyclophosphamide, vincristine, MTX, cytosine arabinoside, and LV). Similarly, following documentation of responses to therapy including MTX,102, 103 among patients with Burkitt’s lymphoma, high-dose MTX with LV rescue has been added to CVAD, cytarabine and intrathecal therapy as well as to other combination chemotherapy regimens104–106 for patients with Burkitt’s lymphoma.
Most treatment regimens for patients with primary CNS lymphoma include high-dose MTX, as conventional radiation doses may lead to neurotoxicity. In a retrospective review of 226 patients with primary CNS lymphoma, those patients treated with regimens that included HDMTX followed by radiotherapy had an improved survival, with no higher risk of late neurotoxicity.107
Choriocarcinoma
Choriocarcinoma is unique in that single-drug treatment with either MTX or actinomycin D produces a substantial number of cures.108 The basis for the unusual sensitivity of this tumor to MTX is not entirely clear, but choriocarcinoma cells may accumulate and retain this drug effectively by synthesizing long-chain polyglutamates. The JAR (human choriocarcinoma) cell line was shown to have active-receptor coupled uptake (potocytosis) of folates and antifolates.109 Current programs for the treatment of this malignancy utilize MTX in combination with other drugs, especially for “poor risk” or relapsed patients.110
Breast cancer
MTX as a single agent causes regressions of breast cancer in approximately 30% of patients. When used with fluorouracil, sequential use of MTX followed by 5-FU improved response rates to 50% and improves disease-free survival when used as adjuvant therapy.111 The adjuvant use of cyclophosphamide, MTX and 5-FU cyclophosphamide methotrexate fluorouracil (CMF) also significantly reduces the risk of relapse,112 may allow more conservative surgery among women with localized disease when used as neoadjuvant therapy,113 and has a role in the treatment of patients with inoperable, advanced disease.114 The combination of MTX, 5-FU with vinorelbine (VMF) instead of cyclophosphamide, has also shown activity among women with advanced breast cancer.115 An additional advantage of this combination is the diminution of long-term toxicity (infertility, carcinogenesis) compared to regimens containing alkylating agents. Finally, it is interesting to note that low-dose oral MTX (2.5 mg BID × 2 days/week) with daily oral cyclophosphamide has shown activity among heavily pretreated women with advanced metastatic breast cancer.116
Gastrointestinal cancer
Antifolates have limited effectiveness in the treatment of gastrointestinal malignancies. The role of MTX in the treatment of these diseases is mainly to modulate, and possibly improve, the effectiveness of 5-FU. By inhibiting purine synthesis, MTX pretreatment increases phosphoribosyl pyrophosphate, a precursor necessary for 5-FU nucleotide formation.117 Data from recent trials using high-dose MTX followed by LV/5-FU in patients with colon cancer highlight the need for a 7- to 24-h interval between MTX and 5-FU administration.118 However, a randomized phase III study comparing 5-FU plus MTX to standard, continuous infusion of 5-FU demonstrated increased toxicity among those treated with MTX, and no difference in overall survival between the two arms. On the basis of these findings, 5-FU remains the standard of care for advanced gastric cancer.119–121
Genitourinary cancer
MTX alone (100 mg/m2), or in high doses (≥500 mg/m2) with LV rescue, is active in the treatment of advanced bladder cancer. The response rate reported (approximately 30%) is similar to the response rate of the other most active single drug, cisplatin. Combinations of drugs including MTX with cisplatin, vinblastine, and doxorubicin (M-VAC) have resulted in a substantial number of long-term clinical remissions.122 A meta-analysis of randomized trials found that neoadjuvant treatment with MTX-containing regimens conferred a survival advantage.123
Head and neck cancer
MTX is an active agent for the treatment of patients with advanced carcinoma of the head and neck region. High-dose MTX regimens with LV rescue appear to improve response rates from 30% to 50%, but remission duration and survival are not improved.124 MTX has also been used with 5-FU in this disease, with response rates as high as 60%.125, 126 The sequence and timing of drug administration have not been shown to affect the response rate, although different patterns of toxicity were observed.
Lung cancer
MTX as a single agent in conventional doses, or in high doses with LV rescue, has only marginal activity in non-small-cell lung cancer (NSCLC). This drug does have limited activity in small cell lung cancer and has been used in combination regimens to treat that disease. Pralatrexate, a nonclassical antifolate discussed later in further detail, has been shown to have activity for patients with previously treated NSCLC.127
Osteogenic sarcoma
Osteosarcoma responds poorly to conventional doses of methotrexate. However, high-dose methotrexate has been one of the mainstays of treatment since its introduction in the 1970s.128 Randomized trials of pre- and post-definitive treatment demonstrated the beneficial effect of chemotherapy that includes high-dose MTX with LV rescue.129 Different studies investigated whether MTX peak concentration and area under concentration–time curve (AUC) are significant prognostic factors of osteosarcoma.130 Controversy remains as to whether there is correlation between MTX exposure and outcome.131 A recent study analyzed the relation between MTX pharmacokinetics, toxicity and survival in children with osteosarcoma. Although there was no correlation between survival and peak concentration or AUC, the 48-h MTX concentration had significant correlation to survival, confirming that systemic MTX exposure was a significant prognostic factor of treatment outcome.132
Neoplastic meningitis
Intrathecal MTX is often a component of therapy for patients with solid-tumor neoplastic meningitis. High-dose IV MTX (8 g/m2) given as the sole treatment with LV rescue may be a reasonable alternative,38 as therapeutic antineoplastic concentrations of MTX can be achieved more easily in the presence of neoplastic meningitis.133
Adverse effects
Hematologic toxicity
Expression of many folate-dependent enzymes targeted by MTX are cell-cycle specific, consistent with their role in DNA synthesis. Tissues that are self-renewing, with a higher S-phase fraction, are therefore at highest risk for damage by the folate antagonists. Bone marrow progenitor cells of all lineages are affected by MTX, but neutropenia usually predominates, with a nadir 10 days after drug administration and recovery typically between days 14 and 21. The effects on the marrow are dose related, but there is considerable variability among patients. Genetic variants,71, 134, 135 subclinical folate deficiency, impaired renal function, a damaged marrow owing to previous radiation therapy, chemotherapy, or infection, and the use of trimethoprim-sulfamethoxazole for Pneumocystis carinii prophylaxis may predispose patients to hematologic (and gastrointestinal) toxicity. Young patients usually tolerate MTX better than older individuals, a fact presumably related to clearance of the drug by the kidneys. The administration of LV can prevent or lessen MTX toxicity and allows larger doses of the antifolate to be administered.
Gastrointestinal toxicity
Nausea and vomiting, even with high doses of MTX, are usually mild to moderate. In contrast, mucositis is a common side effect of MTX treatment. Mucositis usually manifests 3–5 days following exposure to the drug. This is an early sign of MTX toxicity and the drug should be discontinued when it occurs. More severe gastrointestinal toxicity is manifest by diarrhea, which may progress to severe bloody diarrhea. When this occurs in association with neutropenia, patients are at high risk of typhlitis, sepsis, and death. These severe side effects generally occur in a setting of renal damage, usually a consequence of high doses of MTX (≥500 mg/m2/dose), but may also occur in patients treated with conventional doses. MTX blood levels and serum creatinine levels should be followed, and appropriate doses of LV administered, along with supportive measures (see the following discussion).
Renal toxicity
Renal toxicity occurs occasionally following high-dose regimens of MTX and is rare during treatment with lower doses. When it occurs, renal toxicity leads to delayed MTX clearance and subsequently to severe marrow and gastrointestinal toxicity, which can be fatal, especially in adults.136 This toxicity is believed to be due to precipitation of MTX and its less soluble metabolite 7-OH MTX (Figure 3) in the tubules, as well as to a possible direct effect of this drug on the renal tubule.47 The use of vigorous hydration, often with osmotic diuresis and alkalinization of urine to increase solubility of MTX and 7-OH MTX, has markedly ameliorated this problem. Occasional patients, even with this regimen (Table 3), exhibit renal impairment; therefore, careful monitoring of MTX and creatinine serum levels is essential. For those patients who have markedly delayed clearance of MTX secondary to acute renal dysfunction, increasing the dose and rate of LV administration has been the routine therapeutic maneuver to counteract the consequences of prolonged exposure to high MTX levels.137
Table 3 Supportive care for high-dose methotrexate (MTX) treatment
| Pretreatment hydration and alkalinization 8–12 h before treatment, patients should receive 1.5 L/m2 of saline or 5% glucose with 100 mEq HCO3 and 20 mEq KCl/L. Continue hydration until urine pH is 7.0 or greater before MTX administration |
| Monitoring MTX levels should be monitored at 24 h after completion of MTX infusion. Serum creatinine should be measured pretreatment, at 24, and at 48 h |
| Additional LV rescue Required for an MTX level >10−6 M at 24 h. Increase LV dose to 100 mg/m2 q 6 h for levels above 10−6 M and 200 mg/m2 q 6 h for levels above 5 × 10−6 M. Monitor MTX levels daily and continue LV until plasma MTX concentration is <10−8 M |
Abbreviation: LV, leucovorin.
Methylxanthines, such as caffeine or aminophylline, may be useful in the setting of delayed MTX clearance. MTX administration has been shown to increase serum adenosine concentrations, which will decrease glomerular filtration rate.17 Adenosine receptor competitive antagonists, such as the methylxanthines, may therefore act as a specific diuretic to increase MTX elimination.138
Extremely high levels of MTX (>10−3 M) are difficult to rescue, even with high doses of LV.139 Removal of MTX by peritoneal dialysis or hemodialysis is ineffective because the drug is extensively protein bound. Charcoal hemoperfusion columns have been used successfully in a small number of patients, but its efficacy is limited by rebounds in MTX levels.140
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


