Fibrogenic, Fibroosseous, and Fibrohistiocytic Lesions
Fibrogenic, fibroosseous, and fibrohistiocytic lesions of bone represent a clinical spectrum ranging from very innocent lesions requiring no treatment at all to highly malignant neoplasms (36) (Table 4-1). All of these lesions have a common dominant constituent cell, the fibroblast (8,21). In general, fibrous lesions are composed of spindle cells (i.e., fibroclasts and myofibroblasts with characteristic ultrastructural features), which produce a collagenous matrix and so-called ground substance consisting of glycosaminoglycans, whereas fibrohistiocytic lesions may or may not produce a collagenous matrix (35). The term “fibrohistiocytic” is no longer clearly defined. It was introduced for lesions believed to be derived from histiocytes that could act as facultative fibroblasts. Although this concept has been seriously questioned, the term is still in use for descriptive purposes (176).
Although some fibrous lesions, such as nonossifying fibroma (NOF) or periosteal desmoid, are not regarded as tumors or are categorized as miscellaneous lesions by the World Health Organization (WHO), they are included in this text for the purpose of differential diagnosis (14).
Benign Lesions
Fibrous Cortical Defect and Nonossifying Fibroma
Fibrous cortical defect (also called metaphyseal defect) and NOF (formerly called nonosteogenic fibroma) are the most common fibrous lesions of bone and have histologically identical characteristics. Some authors prefer the term fibroxanthoma for both lesions (2,21), whereas Schajowicz (29) preferred the term histiocytic xanthogranuloma. These lesions are not true neoplasms but are considered developmental defects (17), arising in the region of the primary trabeculae of a tubular bone and wandering toward the diaphysis as the bone grows in length (26).
Only two cases of NOF have been so far investigated cytogenetically, revealing a deletion of the short arm of chromosome 4 [edl(4)(p14)] and a clonal chromosomal
translocation t(1;4)(p31:q34) involving the short arm of chromosome 1 and the long arm of chromosome 4 (25,34).
translocation t(1;4)(p31:q34) involving the short arm of chromosome 1 and the long arm of chromosome 4 (25,34).
Table 4-1 Spectrum of Fibrous, Fibroosseous, and Fibrohistiocytic Lesions of Bone | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
Clinical Presentation
Seen predominantly in childhood and adolescence and more commonly (2:1) in boys (5), both lesions have a predilection for the long bones, particularly the femur and the tibia (Fig. 4-1) (8). Fibrous cortical defect is a small, asymptomatic lesion (31) that occurs in 30% of the normal population during the first and second decades of life (7,11,22). The term NOF is applied to this lesion if it enlarges and encroaches on the medullary portion of the bone (28). Depending on age, the distance of these lesions from the growth plate varies (26). When the longitudinal axis of a fibrous metaphyseal defect projects in the direction of the epiphysis it encounters a tendon or ligamentous structure that inserts at the epiphyseal line (27). On the basis of this finding, some investigators believe that such an insertion, along with additional pathogenetic factors, may be related in some degree to the development of this lesion (27). Recent investigations have demonstrated characteristic radiomorphologic stages of progression and regression of fibrous cortical defects (26). At stage A they are located near the growth plate (31), have a round to oval shape, and exhibit a fine sclerotic margin. In stage B the lesion has wandered into the metaphysis and represents a polycyclic form, also delineated by a distinct sclerotic margin, giving the lesion a grape-like appearance. Stage C represents the beginning of the healing process and shows increased mineralization, which characteristically begins at the diaphyseal end of the lesion and progresses toward the growth plate (19,26). Stage D represents complete healing, and the lesion becomes sclerotic.
Most NOFs are asymptomatic, but larger ones may undergo a pathologic fracture (1). Although multifocal presentation is rare (3,12), Moser et al. (24) found that about 8% of patients exhibit a multicentric localization.
The multiple lesions occur in four patterns: (a) clustered together, usually around the knee; (b) nonclustered, at opposite ends of long bones; (c) several small lesions gradually coalescing into one large lesion; and (d) a metachronous appearance, with new lesions eventually appearing near the original lesion (24). Occasionally such multiple NOFs are associated with neurofibromatosis and café-au-lait spots, known as Jaffe-Campanacci syndrome (23,30,32).
The multiple lesions occur in four patterns: (a) clustered together, usually around the knee; (b) nonclustered, at opposite ends of long bones; (c) several small lesions gradually coalescing into one large lesion; and (d) a metachronous appearance, with new lesions eventually appearing near the original lesion (24). Occasionally such multiple NOFs are associated with neurofibromatosis and café-au-lait spots, known as Jaffe-Campanacci syndrome (23,30,32).
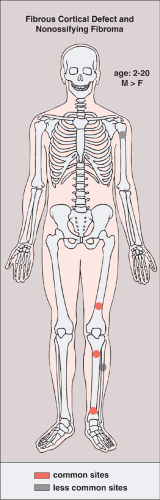 Figure 4-1 Fibrous cortical defect and nonossifying fibroma: skeletal sites of predilection, peak age range, and male-to-female ratio. |
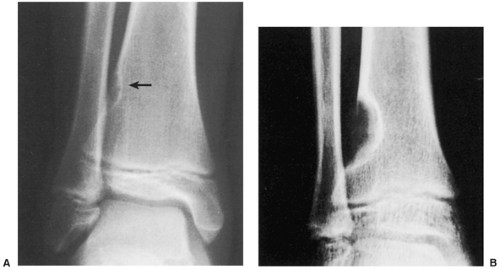 Figure 4-2 Fibrous cortical defect and nonossifying fibroma. A: Fibrous cortical defect, seen here in the lateral cortex of the distal tibia in a 13-year-old boy, typically presents as a radiolucent lesion demarcated by a thin zone of sclerosis (arrow). B: When a fibrous cortical defect encroaches on the medullary cavity, it is called a nonossifying fibroma. Note the similarity of the lesion to that in A. The only difference is the size. |
Imaging
Radiography is usually diagnostic (15). NOFs present as elliptic, radiolucent defects confined to the cortex of a long bone near the growth plate, frequently resembling a bunch of grapes and demarcated by a thin sclerotic margin (Fig. 4-2). They range in size from 0.5 to 7 cm, with their long axes aligned with the long axis of the affected bone (17). The larger defects are usually scalloped and have a wider zone of sclerosis. Most foci undergo spontaneous involution (healing) by the remodeling process of the tubular bone with diaphyseal diminution of the diameter finally resulting in osteosclerosis. A few lesions may continue to grow and as they encroach on the medullary cavity they characteristically display a scalloped, sclerotic border and are located eccentrically within the bone (Figs. 4-3 and 4-4). Some of the larger lesions may be complicated by a fracture (Fig. 4-5).
Skeletal scintigraphy shows a minimal to mild increase in activity (4). During the healing phase, mild hyperemia may be seen on the blood pool image, and the positive delayed scan reflects the osteoblastic activity (16). Computed tomography (CT) may demonstrate to better advantage the cortical thinning and medullary involvement (Fig. 4-6) and may more precisely delineate early pathologic fracture. Hounsfield attenuation values for NOF are higher than for normal bone marrow. Magnetic resonance imaging (MRI), usually performed for another reason, shows a low signal intensity on
T1-weighted sequences (17) (Figs. 4-7 and 4-8B). On T2 weighting the lesion exhibits either low signal (more common) or high signal intensity (less common) (20) (Fig. 4-8C). After gadolinium diethylenetriamine–penta-acetic (DTPA) injection, NOFs invariably exhibit a hyperintense border and signal enhancement (27) (Fig. 4-8D). Mineralization of the lesion during healing (stages C and D) appears predominantly as low signal intensity on MR images.
T1-weighted sequences (17) (Figs. 4-7 and 4-8B). On T2 weighting the lesion exhibits either low signal (more common) or high signal intensity (less common) (20) (Fig. 4-8C). After gadolinium diethylenetriamine–penta-acetic (DTPA) injection, NOFs invariably exhibit a hyperintense border and signal enhancement (27) (Fig. 4-8D). Mineralization of the lesion during healing (stages C and D) appears predominantly as low signal intensity on MR images.
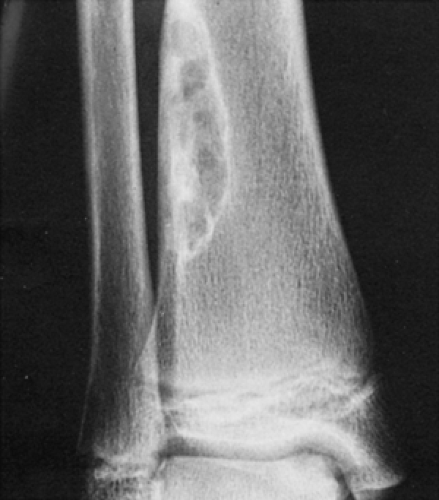 Figure 4-3 Nonossifying fibroma. The lesion, seen here in the distal tibia in an asymptomatic 15-year-old boy, appears eccentrically located in the bone and has a scalloped sclerotic border. |
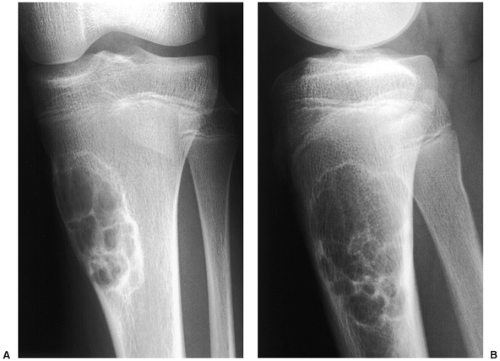 Figure 4-4 Nonossifying fibroma. A, B: Anteroposterior and lateral radiographs show a large elliptical, lobulated lesion in the proximal tibial diaphysis exhibiting well-defined sclerotic border. Note the eccentric location of the lesion, which only slightly scallops the endosteal aspect of the anteromedial cortex. |
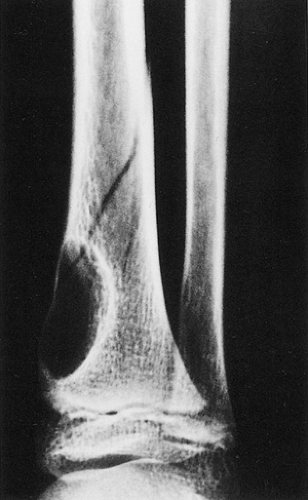 Figure 4-5 Nonossifying fibroma: a pathologic fracture. A large radiolucent lesion in the distal tibia of a 10-year-old boy is complicated by a pathologic fracture. |
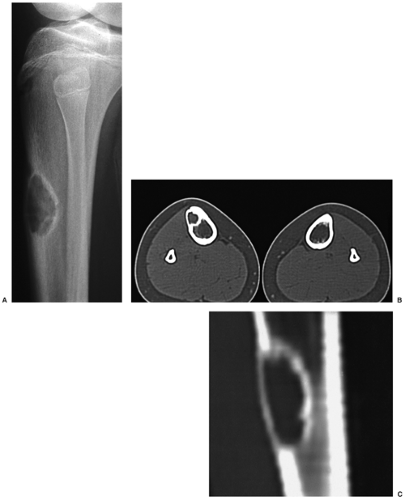 Figure 4-6 Nonossifying fibroma (NOF): computed tomography (CT). A: Oblique radiograph of the right tibia of a 14-year-old girl shows an elliptical radiolucent lesion with sclerotic border. B: Axial CT section shows the intramedullary lesion with well-defined sclerotic border extending into the anterior cortex of the tibia. C: Coronal reformatted CT shows the full extent of NOF. Observe thinning of the anterior cortex. |
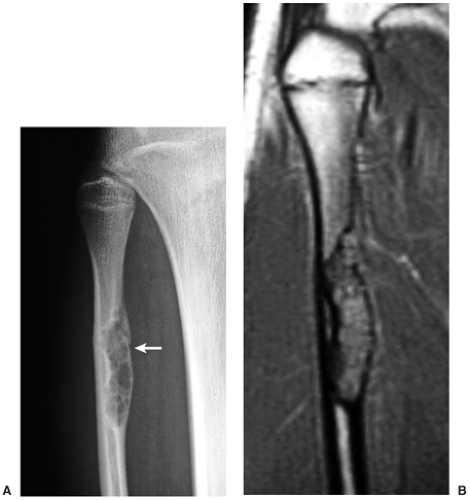 Figure 4-7 Nonossifying fibroma: magnetic resonance imaging (MRI). A: Anteroposterior radiograph of the right fibula of a 14-year-old girl shows an eccentric well-defined radiolucent lesion with sclerotic border. Note thinning of the medial cortex and a pathologic fracture (arrow). B: Coronal T1-weighted MRI shows the lesion exhibiting low signal intensity. |
Histopathology
Histologically identical regardless of size, fibrous cortical defect and NOF are composed of rather bland monomorphous spindle cells admixed with histiocytic cells that often possess a clear or foamy cytoplasm (33). The cells are frequently arranged in a swirling storiform pattern (derived from Greek and implying resemblance to a woven mat), typical of fibrohistiocytic lesions, but a fascicular arrangement may also be seen (Fig. 4-9). A variable number of lipid-bearing xanthomatous cells and of hemosiderin pigment–laden histiocytes may be present (29) (see Fig. 4-9C). In some cases, unusually heavily lipidized fibroxanthomatous areas are predominant. Such lesions have been referred to as xanthoma or fibroxanthoma by some investigators (2). Xanthomas in bone may also rarely be associated with juvenile xanthogranuloma or xanthoma disseminatum, a cutaneous form of non–Langerhans cell histiocytosis, and with hyperlipidemia, usually found at older ages and in atypical skeletal sites, such as flat bones, or as diaphyseal lesions (9,10,13). In hyperlipidemia, cutaneous xanthelasmas and detectable disturbances in lipid values may be absent at the time of bone manifestations (9). Osteoclast-like giant cells are usually present in NOF (Fig. 4-10) and, when numerous, may lead to confusion with giant cell tumor of bone (18). Typically, variable numbers of inflammatory cells, especially lymphocytes, and a few plasma cells are scattered in the background. The amount of collagen production may vary, but no bone or cartilage formation should be present unless a fracture or previous surgical disruption of the lesion has occurred. Exceptionally, bizarre pseudosarcomatous nuclear features may be regarded as degenerative or ischemic changes that may raise the suspicion of malignancy when radiologic and clinical details are not taken into consideration (6).
Differential Diagnosis
Radiology
Both fibrous cortical defect and NOF have rather characteristic imaging features (see previous discussion) that make radiography diagnostic. Rarely, a fibrous cortical defect affecting the posterior aspect of the distal
femur may be mistaken for periosteal desmoid, and a NOF in long or flat bones may be mistaken for a focus of fibrous dysplasia or desmoplastic fibroma. An atypical sclerotic presentation of NOF or healing stage of this lesion may occasionally be confused with a medullary bone infarct, sclerotic metastasis, or a giant bone island (Fig. 4-11). If a lesion is located in thin bones (e.g., fibula, ulna) and occupies the entire diameter of the bone, its characteristic eccentric location is lost and it may therefore mimic a simple bone cyst (Fig. 4-12), an aneurysmal bone cyst (Fig. 4-13), or a Langerhans cell histiocytosis. Whenever NOF affects the anterior aspect of the tibia, osteofibrous dysplasia should be considered in differential diagnosis (Fig. 4-14).
femur may be mistaken for periosteal desmoid, and a NOF in long or flat bones may be mistaken for a focus of fibrous dysplasia or desmoplastic fibroma. An atypical sclerotic presentation of NOF or healing stage of this lesion may occasionally be confused with a medullary bone infarct, sclerotic metastasis, or a giant bone island (Fig. 4-11). If a lesion is located in thin bones (e.g., fibula, ulna) and occupies the entire diameter of the bone, its characteristic eccentric location is lost and it may therefore mimic a simple bone cyst (Fig. 4-12), an aneurysmal bone cyst (Fig. 4-13), or a Langerhans cell histiocytosis. Whenever NOF affects the anterior aspect of the tibia, osteofibrous dysplasia should be considered in differential diagnosis (Fig. 4-14).
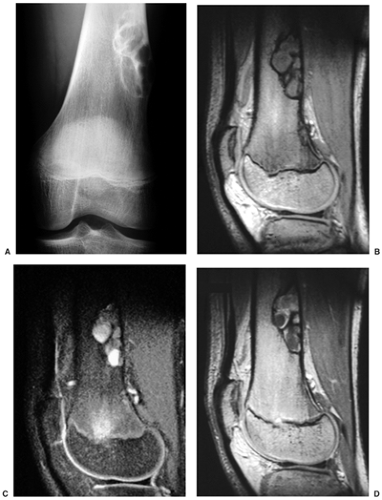 Figure 4-8 Nonossifying fibroma (NOF): magnetic resonance imaging (MRI). A: Anteroposterior radiograph shows a radiolucent lesion with sclerotic border abutting the posteromedial cortex of the right femur. B: Sagittal T1-weighted MRI shows predominantly low signal intensity of the lesion. C: Sagittal T2-weighted image shows heterogeneous but mostly high signal intensity of NOF. D: Sagittal T1-weighted fat-suppressed MRI after intravenous injection of gadolinium shows slight heterogeneous enhancement of the lesion. |
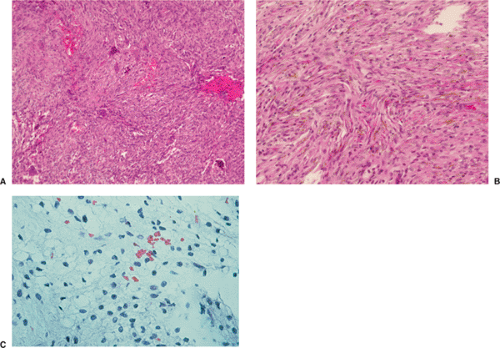 Figure 4-9 Histopathology of nonossifying fibroma. A: Fibrous stroma displays a storiform arrangement. Several giant cells are present (hematoxylin and eosin, original magnification ×100). B: The fibrous stroma exhibits a storiform arrangement of slender cells (hematoxylin and eosin, original magnification ×200). C: At high magnification several foam cells are conspicuous (hematoxylin and eosin, original magnification ×400). |
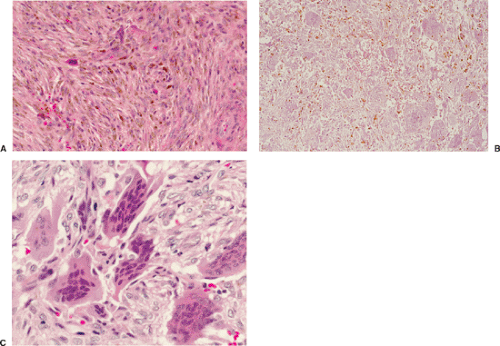 Figure 4-10 Histopathology of nonossifying fibroma. A: Siderin-containing macrophages are irregularly distributed within a fairly dense spindle cell tissue (Giemsa, original magnification ×200). B: In another area the number and the size of the giant cells containing up to 50 nuclei resemble that of a true giant cell tumor of bone (hematoxylin and eosin, original magnification ×50). C: At high magnification the resemblance of giant cells to those of a giant cell tumor is striking (hematoxylin and eosin, original magnification ×400). |
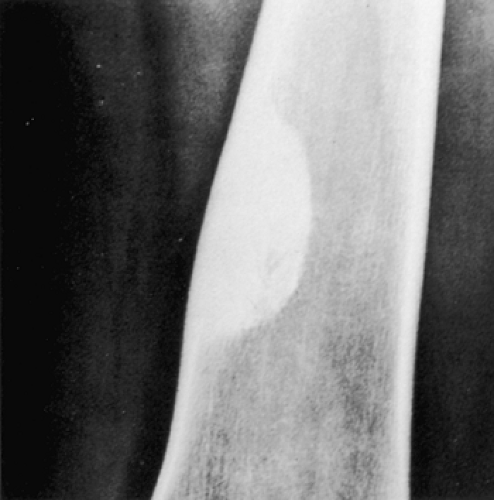 Figure 4-11 Healed nonossifying fibroma. A lesion that healed spontaneously may persist as a sclerotic patch. In this sclerosing phase, nonossifying fibroma should not be mistaken for osteoblastic tumor or bone island. |
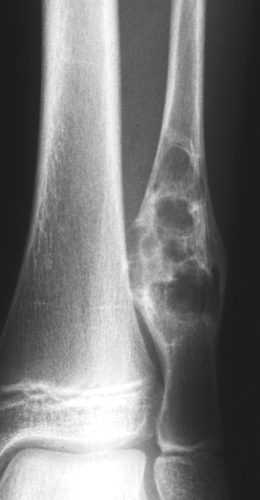 Figure 4-12 Nonossifying fibroma resembling a simple bone cyst. Multiloculated lesion in the distal fibula mimics a simple bone cyst. Periosteal reaction at the lateral cortex is secondary to a healed pathologic fracture. |
Pathology
NOF can be confused with benign fibrous histiocytoma because both lesions have almost identical histopathologic features, differing mainly in their clinical and radiologic presentation. A storiform architecture in a hypercellular NOF or pseudosarcomatous nuclear changes can lead to confusion with malignant fibrous histiocytoma (MFH) or unclassified sarcoma if the pathologist is unfamiliar with the clinicopathologic spectrum of fibrous cortical defect and NOF. Less likely differential possibilities are giant cell tumor, fibrous dysplasia, osteofibrous dysplasia, or low-grade intraosseous osteosarcoma. Although giant cells are almost always present in NOF, they are not uniformly distributed throughout the lesion as in giant cell tumor (see Figs. 4-10 and 7-10). Because bone formation is not a feature of NOF, all three lesions—fibrous dysplasia, osteofibrous dysplasia, and low-grade osteosarcoma—can easily be distinguished. If xanthoma cells are numerous and a storiform pattern of fibrohistiocytic cells is not obvious, disturbances of lipid metabolism (in elderly patients) or non–Langerhans cell histiocytic disorders (in children or adolescents, ordinarily involving also the dermis) with osseous manifestations must be considered.
The radiologic and pathologic differential diagnosis of NOF is depicted in Figure 4-15.
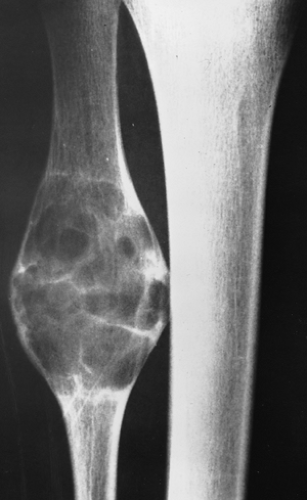 Figure 4-13 Nonossifying fibroma resembling an aneurysmal bone cyst. A large lesion in the fibula exhibits an expansive character and thus resembles an aneurysmal bone cyst. Note, however, the absence of a periosteal reaction that is invariably present in the latter lesion. |
Benign Fibrous Histiocytoma
The term benign fibrous histiocytoma (sometimes called xanthofibroma or fibrous xanthoma) may be controversial (36,42,47), but it is useful in an organizational sense for subclassification of lesions with histologic features similar to those of NOF but having an atypical location and an atypical clinical or radiographic pattern (45,46,48). This is a rare lesion characterized by a spindle cell fibrous tissue arranged in a storiform pattern. The tissue contains variable numbers of giant cells, hemosiderin, and lipid-laden histiocytes (45).
Clinical Presentation
The lesion, which is often symptomatic (painful), is usually diagnosed in older patients who are in the age range from 15 to 60 years. The usual location is the shaft or (in contrast to NOF) the articular end of a long bone, the pelvis, and the ribs. Rarely, the clavicle, spine, or skull is affected (29) (Fig. 4-16). Clinically, the lesion can exhibit a locally aggressive behavior, with recurrences after curettage or excision.
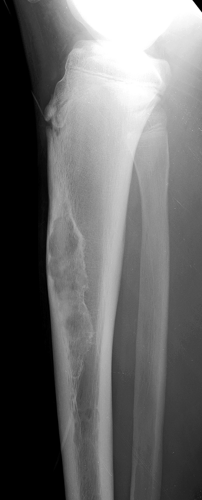 Figure 4-14 Nonossifying fibroma resembling osteofibrous dysplasia. Lateral radiograph of the leg shows a long lesion with sclerotic border affecting the anterior cortex of the tibia and extending into the medullary portion of bone. Because this is a preferential location for osteofibrous dysplasia, the latter diagnosis must be considered. |
Imaging
Radiographically, benign fibrous histiocytoma may also appear to be more aggressive than NOF, although some features are similar. Radiographs show a well-defined, radiolucent lesion with sclerotic borders, occasionally exhibiting some degree of expansion (41) (Fig. 4-17). Internal trabeculations may also be present (38). The lesion may be situated either centrally in the bone or eccentrically, the latter presentation being more common at the articular end of bone (43). On skeletal scintigraphy, benign fibrous histiocytoma exhibits a moderately increased uptake (41). MRI shows the lesion to be of intermediate signal intensity (isointense with the muscles) on spin-echo (SE) T1-weighted sequences and of a high signal intensity on T2- and inversion time–inversion recovery (TI-IR) images (37,41).
Histopathology
Benign fibrous histiocytoma usually has histologic features quite similar to those of NOF, although the storiform spindle cell stroma is more distinctive (29,44)
(Fig. 4-18). The background may contain larger numbers of histiocytic cells and Touton-type giant cells seen in xanthomatous tissue reactions (39) or in MFH. Although frankly malignant histologic criteria are absent and no cytologic atypia is seen, mitotic activity may be present (40). Reactive bone formation may be seen at the periphery of the lesion or in association with a pathologic fracture through the lesion (36).
(Fig. 4-18). The background may contain larger numbers of histiocytic cells and Touton-type giant cells seen in xanthomatous tissue reactions (39) or in MFH. Although frankly malignant histologic criteria are absent and no cytologic atypia is seen, mitotic activity may be present (40). Reactive bone formation may be seen at the periphery of the lesion or in association with a pathologic fracture through the lesion (36).
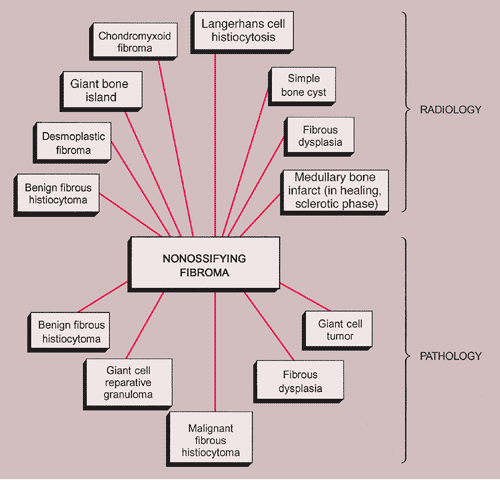 Figure 4-15 Radiologic and pathologic differential diagnosis of nonossifying fibroma. |
Differential Diagnosis
Radiology
From the radiologic standpoint, the main differential possibilities include NOF and giant cell tumor, particularly when benign fibrous histiocytoma is situated at the articular end of bone. The radiographic features of NOF may be indistinguishable from those of benign fibrous histiocytoma, although the more aggressive appearance of the latter (such as an expanding border) and the location, which is atypical for NOF, are the clues to the diagnosis. Giant cell tumor only rarely exhibits a rim of reactive sclerosis, whereas this is a common feature of benign fibrous histiocytoma. Although intraosseous ganglion is usually much smaller than the average size of benign fibrous histiocytoma, the former should be considered in differential diagnosis if the lesion occurs at the articular end of a bone. Osteoblastoma, unlike benign fibrous histiocytoma, usually evokes a periosteal reaction and exhibits central opacities of bone formation.
Pathology
The differentiation of NOF and fibrous cortical defect from benign fibrous histiocytoma must be made on a clinical and radiographic basis rather than on histologic grounds (36) because the microscopic appearance of all three lesions is almost identical (Fig. 4-19).
Giant cell tumor always must be considered in the differential diagnosis. It is particularly difficult to distinguish a giant cell tumor that undergoes regressive changes and, conversely, some giant cell tumors with regressive features may erroneously be diagnosed as benign fibrous histiocytoma (38). Furthermore, the healing reaction associated with a pathologic fracture in a giant cell tumor can lead to proliferation of spindle cells with associated lipid- and hemosiderin-laden macrophages, giving the appearance of benign fibrous histiocytoma (36). Finally, some investigators consider benign fibrous histiocytoma a regressive stage of a burned-out giant cell tumor (43).
The radiologic and pathologic differential diagnosis of benign fibrous histiocytoma is depicted in Figure 4-20.
Periosteal Desmoid
Periosteal desmoid (also known as avulsive cortical irregularity, distal metaphyseal femoral defect, cortical desmoid, or medial supracondylar defect of the femur) (52) is a tumor-like fibrous proliferation of
the periosteum, which has a striking predilection for the posteromedial cortex of the medial femoral condyle (50). It was first described in 1951 by Kimmelstiel and Rapp (54) as a benign fibrous lesion originating beneath the periosteum and associated with erosion of the underlying bone. Although the term periosteal desmoid is widely accepted, it has an unfortunate similarity to the term desmoid tumor of soft tissue and therefore is suggestive of an aggressive lesion (35). Periosteal desmoid usually occurs between the ages of 12 and 20 years, mainly in boys, and most lesions disappear spontaneously by the end of the second decade. It is considered to represent an asymptomatic normal variant that does not require biopsy or treatment. Although many patients have a history of injury, trauma does not necessarily predispose to the development of this lesion (56). The radiologic features of periosteal desmoid are similar to those of fibrous cortical defect, except for the specificity of its location. Its radiographic hallmarks are a saucer-like radiolucent defect with sclerosis at its base. The lesion may erode the cortex (55) and appear as a cortical irregularity (Fig. 4-21). Occasionally, because of its poorly defined and irregular bony margin and the presence of small bony spiculae at the cortical surface, it may simulate an aggressive or even a malignant tumor, such as osteosarcoma or Ewing sarcoma (49,50). The bone scan usually is normal but sometimes may show a focal increase in activity (17,53,57). On MRI the lesion appears hypointense on T1- and hyperintense on T2-weighted images, with a dark rim on both sequences at or near the sites of the bone attachment of the medial head of the gastrocnemius muscle (58). Histopathologic examination of the lesion demonstrates fibroblastic spindle cells that produce large amounts of collagen (51)
(Fig. 4-22). Large areas of hyalinization and fibrocartilage and small fragments of bone may be scattered within the fibrous tissue. Newly formed capillaries may also be present. Unlike NOF or fibrous cortical defect, giant cells or xanthomatous cells are not present. The most important point in the differential diagnosis is not to mistake this lesion for a malignancy. Its characteristic radiographic appearance and location should serve as clues to the correct diagnosis.
the periosteum, which has a striking predilection for the posteromedial cortex of the medial femoral condyle (50). It was first described in 1951 by Kimmelstiel and Rapp (54) as a benign fibrous lesion originating beneath the periosteum and associated with erosion of the underlying bone. Although the term periosteal desmoid is widely accepted, it has an unfortunate similarity to the term desmoid tumor of soft tissue and therefore is suggestive of an aggressive lesion (35). Periosteal desmoid usually occurs between the ages of 12 and 20 years, mainly in boys, and most lesions disappear spontaneously by the end of the second decade. It is considered to represent an asymptomatic normal variant that does not require biopsy or treatment. Although many patients have a history of injury, trauma does not necessarily predispose to the development of this lesion (56). The radiologic features of periosteal desmoid are similar to those of fibrous cortical defect, except for the specificity of its location. Its radiographic hallmarks are a saucer-like radiolucent defect with sclerosis at its base. The lesion may erode the cortex (55) and appear as a cortical irregularity (Fig. 4-21). Occasionally, because of its poorly defined and irregular bony margin and the presence of small bony spiculae at the cortical surface, it may simulate an aggressive or even a malignant tumor, such as osteosarcoma or Ewing sarcoma (49,50). The bone scan usually is normal but sometimes may show a focal increase in activity (17,53,57). On MRI the lesion appears hypointense on T1- and hyperintense on T2-weighted images, with a dark rim on both sequences at or near the sites of the bone attachment of the medial head of the gastrocnemius muscle (58). Histopathologic examination of the lesion demonstrates fibroblastic spindle cells that produce large amounts of collagen (51)
(Fig. 4-22). Large areas of hyalinization and fibrocartilage and small fragments of bone may be scattered within the fibrous tissue. Newly formed capillaries may also be present. Unlike NOF or fibrous cortical defect, giant cells or xanthomatous cells are not present. The most important point in the differential diagnosis is not to mistake this lesion for a malignancy. Its characteristic radiographic appearance and location should serve as clues to the correct diagnosis.
 Figure 4-16 Benign fibrous histiocytoma: skeletal sites of predilection, peak age range, and male-to-female ratio. |
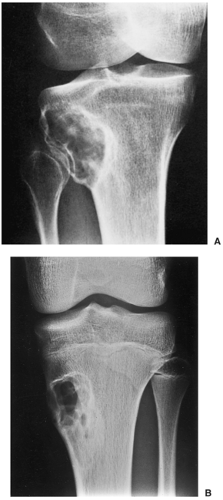 Figure 4-17 Benign fibrous histiocytoma. A: A 37-year-old man presented with occasional pain in the right knee. An oblique radiograph of the knee shows a lobulated radiolucent lesion with a well-defined sclerotic border, located eccentrically in the proximal tibia. B: A 16-year-old boy presented with a painful tibial lesion that on radiography looked like a nonossifying fibroma. Because of persistent symptoms the lesion was biopsied and proved to be consistent with a benign fibrous histiocytoma. |
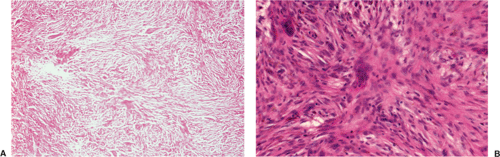 Figure 4-18 Histopathology of benign fibrous histiocytoma. A: Benign-appearing fibrous tissue displays a storiform arrangement (hematoxylin and eosin, original magnification ×100). B: Giant cells and some inflammatory cells are present. The spindle cells do not exhibit atypical features (hematoxylin and eosin, original magnification ×200). |
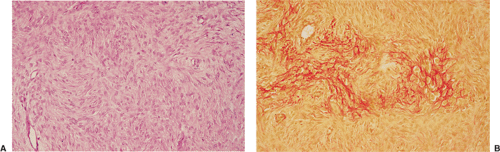 Figure 4-19 Histopathology of benign fibrous histiocytoma. A: Densely arranged spindle cells exhibit a cartwheel-like arrangement (center). In contrast to nonossifying fibroma, giant cells are rarely encountered (hematoxylin and eosin, original magnification ×50). B: Spotty increase of collagen fiber formation (red) resembles primitive osteoid (van Gieson, original magnification ×20). |
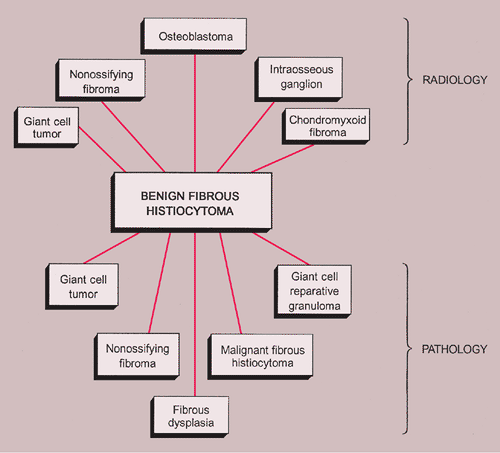 Figure 4-20 Radiologic and pathologic differential diagnosis of benign fibrous histiocytoma. |
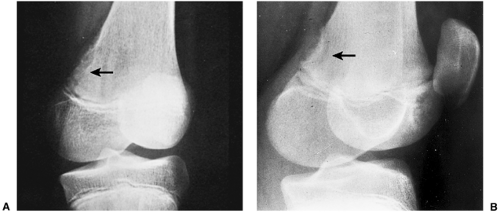 Figure 4-21 Periosteal desmoid. A: Oblique radiograph of the left knee of a 12-year-old boy shows the classic appearance of periosteal desmoid. Note the elliptical radiolucency eroding the medial border of the distal femoral metaphysis at the linea aspera and producing cortical irregularity (arrow). B: In another patient, an 11-year-old boy, a saucer-like defect is present in the medial cortex of the distal femoral metaphysis (arrow). |
The radiologic and pathologic differential diagnosis of periosteal desmoid is shown in Figure 4-23.
Fibrous Dysplasia
Fibrous dysplasia, occasionally termed fibrous osteodystrophy, osteodystrophia fibrosa, and osteitis fibrosa disseminata (70), is a fibroosseous lesion that may affect one bone (monostotic form, 70% to 80% of patients) or
several bones (polyostotic form, 20% to 30%) (98). Classified as a developmental abnormality by some authorities, it is now considered as a genetically based sporadic disorder and is characterized by the replacement of normal lamellar cancellous bone by an abnormal fibrous tissue that contains small, abnormally arranged trabeculae of immature woven bone (17,91,118), formed by metaplasia of the fibrous stroma. Fibrous dysplasia presents with an equal gender distribution and occurs in all ethnic groups. Although monostotic fibrous dysplasia is usually diagnosed in young adults and polyostotic forms in children and adolescents, syndrome-associated lesions may be detected as early as in infancy (118).
several bones (polyostotic form, 20% to 30%) (98). Classified as a developmental abnormality by some authorities, it is now considered as a genetically based sporadic disorder and is characterized by the replacement of normal lamellar cancellous bone by an abnormal fibrous tissue that contains small, abnormally arranged trabeculae of immature woven bone (17,91,118), formed by metaplasia of the fibrous stroma. Fibrous dysplasia presents with an equal gender distribution and occurs in all ethnic groups. Although monostotic fibrous dysplasia is usually diagnosed in young adults and polyostotic forms in children and adolescents, syndrome-associated lesions may be detected as early as in infancy (118).
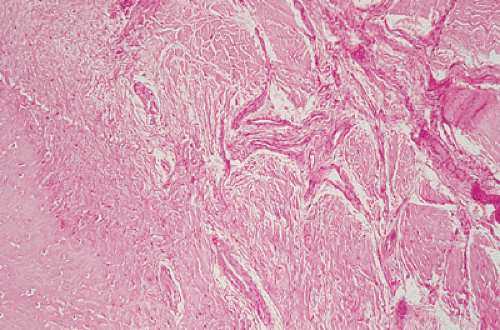 Figure 4-22 Histopathology of periosteal desmoid. The cortical bone (lower left) is made up of coarse plates of cell-rich woven bone merging with benign-appearing fibrous tissue (hematoxylin and eosin, original magnification ×12). |
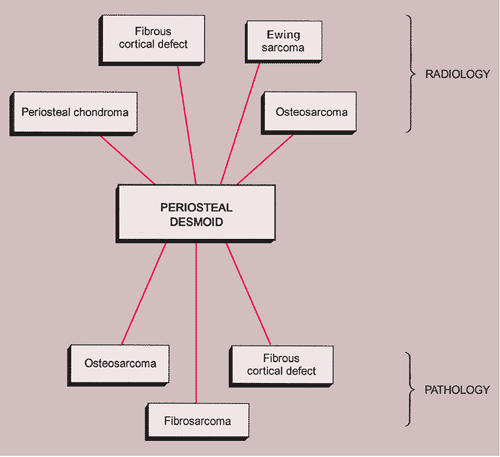 Figure 4-23 Radiologic and pathologic differential diagnosis of periosteal desmoid. |
A mutation in the GNAS1 gene that codes for the alpha-subunit of a signal transducing membrane protein (Guanine Nucleotide-finding Protein, Alpha-Stimulating Activity Polypeptide 1) is involved in fibrous dysplasia. This protein belongs to the group of guanine nucleotide binding proteins, or G-proteins, involved in second messenger cascades. Because mutations in this autosomal gene located on 20q13 are lethal, only postzygotic mutations leading to mosaicism, i.e., to the existence of two cell lines with a different genetic makeup within one individual, are observed (81,82). Depending on whether it occurs during embryonic development or postnatal life, it is believed that the mutation will lead to monostotic or polyostotic fibrous dysplasia or to McCune-Albright syndrome. The GNAS1 mutation is of the activating type, causing increased activity of the enzyme adenylcyclase, which leads to overproduction of cyclic adenosine monophosphate (cAMP) (117). This affects proliferation and differentiation of preosteoblasts by increasing the expression of genes that contain cAMP-responsive elements, such as c-fos, c-jun, IL-6, and Il-11 (65,67,101,102,113).
Clonal chromosomal alterations have been reported in 8 of 11 investigated cases of monostotic fibrous dysplasia, with structural recurrent aberrations of chromosome
12 (12p13) and trisomy 2 in three cases each, suggesting that fibrous dysplasia may be a neoplastic process (73).
12 (12p13) and trisomy 2 in three cases each, suggesting that fibrous dysplasia may be a neoplastic process (73).
Monostotic Fibrous Dysplasia
Clinical Presentation
Most commonly affecting the femur (with a predilection for the femoral neck), the tibia, the ribs (representing the most common benign lesion of the rib), and the base of the skull, the lesion of monostotic fibrous dysplasia arises centrally in the bone; it usually spares the epiphysis in children and the articular end of the bone in adults (83). The incidence in men and women is the same (Fig. 4-24). Because these lesions are usually asymptomatic, most of them are discovered incidentally on radiographs obtained for other reasons. Rarely, pain accompanied by local swelling and deformity is present. The most common complication is a pathologic fracture through the structurally weakened bone. This may be the initial presentation, particularly for lesions located in the lower extremity (29).
Imaging
The radiographic appearance of the lesion depends on the proportion of osseous to fibrous tissue. Lesions with higher degrees of ossification appear more dense and sclerotic. More fibrous lesions exhibit a greater radiolucency, with a characteristic “ground-glass” or even cystic appearance (Fig. 4-25). Some lesions may contain scattered calcifications similar to those of chondroid lesions. A solitary focus of fibrous dysplasia may be surrounded by a characteristic thick band of reactive bone, creating a “rind” sign (Fig. 4-26). This appearance of fibrous dysplasia must be differentiated from the very similar presentation of a bone infarct (see Fig. 4-43B). When the lesion enlarges, it expands the medullary cavity. Although the cortex may be attenuated and even remodeled around the lesion, it is rarely directly involved by the lesion. In extremely rare instances, lesions of fibrous dysplasia may protrude from the bone (the so-called exophytic variant of fibrous dysplasia) (75).
Scintigraphy is helpful in determining the activity (Fig. 4-27) and the potential multicentricity of the lesion (86). Machida et al. (100) reported that although a high incidence of increased uptake of radiopharmaceutical was seen in 59 patients with fibrous dysplasia, 10% of the lesions with a ground-glass appearance failed to show similarly increased uptake.
The lesion of fibrous dysplasia shows a variety of appearances on MRI (94). Some lesions show a decreased signal on both T1 and T2 sequences and some low signal on T1 but either mixed or high signal on T2 images (90). The sclerotic rim (rind sign) is invariably imaged as a band of low signal intensity on both T1 and T2 sequences (122).
Histopathology
Histologically, fibrous dysplasia consists of an aggregate of moderately dense fibrous connective tissue composed
of spindle cells arrayed in intersecting, whorled bundles containing haphazardly distributed bone trabeculae. Rather than being stress oriented, as in normal cancellous bone, these trabeculae tend to be irregularly curved and branching with sparse interconnections (62,107). The collagen fibers of the stroma are characteristically continuous with those of the bone trabeculae, reminiscent of Sharpey fibers. The low-power photomicrographic picture has been likened to Chinese characters or even alphabet soup (Fig. 4-28). Although bone trabeculae contain osteocytes, they exhibit no evidence of osteoblastic activity. In some areas, cementicles-like rounded trabeculae may be present (Fig. 4-29) and scattered osteoclasts may be found. Areas of hyaline cartilage may rarely be present. Under polarized light, the bone trabeculae are seen to be composed of woven immature bone (Fig. 4-30). The cells of the stroma and of the trabeculae show a strong alkaline phosphatase activity, indicating that they are all potential osteoblasts. It appears that an organizational defect, now identified as mutation of the GNAS1 gene, prevents them from differentiating into fully mature, polarized osteoblasts capable of forming normal lamellar bone. The lack of seams of polarized osteoblasts at any stage in their maturation (“naked trabeculae”) and the direct passage of the collagen fibers from the stroma into the bone trabeculae are the most important characteristics in differential diagnosis (see Fig. 4-29). Therefore, the bone formed is considered immature (see Fig. 4-30). Furthermore, typical findings on electron microscopy consist of fairly extended areas with collagen fibrils in the precollagen stage and in continuation with normal-appearing mature collagen fibrils and calcification (Remagen, unpublished observations, 1996) (Fig. 4-31).
of spindle cells arrayed in intersecting, whorled bundles containing haphazardly distributed bone trabeculae. Rather than being stress oriented, as in normal cancellous bone, these trabeculae tend to be irregularly curved and branching with sparse interconnections (62,107). The collagen fibers of the stroma are characteristically continuous with those of the bone trabeculae, reminiscent of Sharpey fibers. The low-power photomicrographic picture has been likened to Chinese characters or even alphabet soup (Fig. 4-28). Although bone trabeculae contain osteocytes, they exhibit no evidence of osteoblastic activity. In some areas, cementicles-like rounded trabeculae may be present (Fig. 4-29) and scattered osteoclasts may be found. Areas of hyaline cartilage may rarely be present. Under polarized light, the bone trabeculae are seen to be composed of woven immature bone (Fig. 4-30). The cells of the stroma and of the trabeculae show a strong alkaline phosphatase activity, indicating that they are all potential osteoblasts. It appears that an organizational defect, now identified as mutation of the GNAS1 gene, prevents them from differentiating into fully mature, polarized osteoblasts capable of forming normal lamellar bone. The lack of seams of polarized osteoblasts at any stage in their maturation (“naked trabeculae”) and the direct passage of the collagen fibers from the stroma into the bone trabeculae are the most important characteristics in differential diagnosis (see Fig. 4-29). Therefore, the bone formed is considered immature (see Fig. 4-30). Furthermore, typical findings on electron microscopy consist of fairly extended areas with collagen fibrils in the precollagen stage and in continuation with normal-appearing mature collagen fibrils and calcification (Remagen, unpublished observations, 1996) (Fig. 4-31).
 Figure 4-24 Monostotic fibrous dysplasia: skeletal sites of predilection, peak age range, and male-to-female ratio. |
 Figure 4-25 Monostotic fibrous dysplasia. A: Anteroposterior radiograph of the distal leg of a 17-year-old girl shows a monostotic focus of fibrous dysplasia in the diaphysis of the tibia. Observe the slight expansion and thinning of the cortex and the partial loss of trabecular pattern in the cancellous bone, which gives the lesion a “ground-glass” or “smoky” appearance. B: Anteroposterior radiograph of the right hip in a 25-year-old man shows the focus of fibrous dysplasia in the femoral neck that exhibits a more sclerotic appearance than that seen in A. |
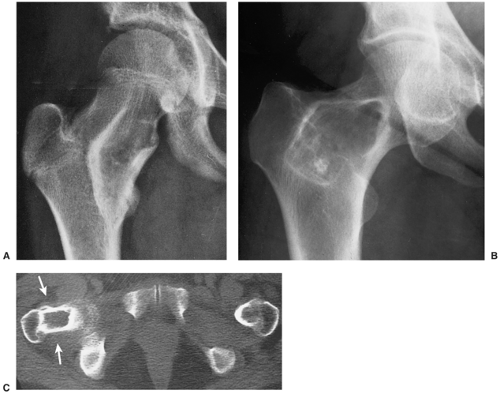 Figure 4-26 Monostotic fibrous dysplasia. A: In this presentation of fibrous dysplasia the thick peripheral rim and a radiolucent center create the image of a “rind” sign. B: In another patient a similar rind sign marks a focus of fibrous dysplasia. C: Computed tomography section through the lesion shows a thick rim of peripheral sclerosis (arrows). |
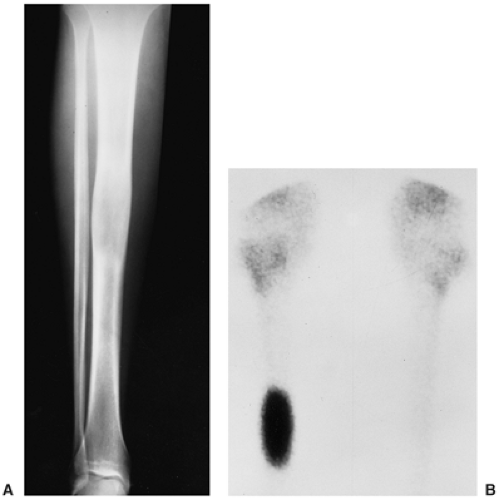 Figure 4-27 Monostotic fibrous dysplasia: scintigraphy. A: A 24-year-old woman presented with mild discomfort in the right leg. Anteroposterior radiograph shows a radiolucent lesion with a “smoky” appearance in the midshaft of the tibia, associated with thinning of the cortex and slight expansion. B: Radionuclide bone scan shows markedly increased uptake of the radiopharmaceutical agent, indicating metabolic activity of fibrous dysplasia. |
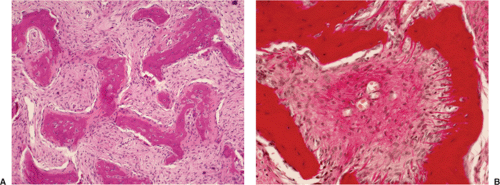 Figure 4-28 Histopathology of fibrous dysplasia. A: Metaplastic trabeculae are haphazardly arranged in loose, fibrous stroma. Note lack of osteoblastic activity (hematoxylin and eosin, original magnification ×200). B: Collagen, reminiscent of Sharpey fibers, extends into the stroma surrounding the trabeculae. Note the absence of osteoblastic rimming (van Gieson, original magnification ×200) |
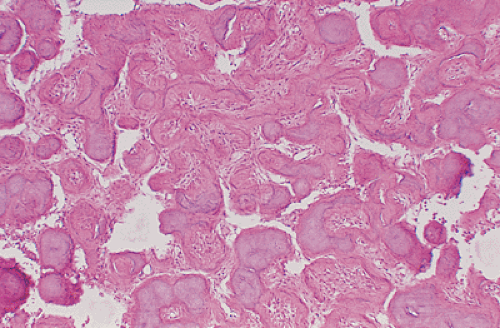 Figure 4-29 Histopathology of fibrous dysplasia. Variant of classic histologic appearance with small, discrete, almost osteocyte-free foci of calcified matrix that resemble the cementicles usually seen in cementifying fibromas of the jaw. The scanty spindle cell stroma shows the typical passing of the collagen fibers into the bone (hematoxylin and eosin, original magnification ×25). |
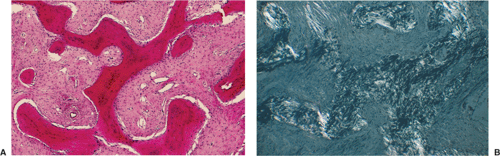 Figure 4-30 Histopathology of fibrous dysplasia. A: In a background of collagenized fibrous tissue, irregularly shaped spicules of immature woven bone have been formed. No polarized differentiated osteoblasts are seen (hematoxylin and eosin, original magnification ×50). B: Under polarized light the woven structure of the collagen fibers within the bone matrix is conspicuous (hematoxylin and eosin, polarized light, original magnification ×25). |
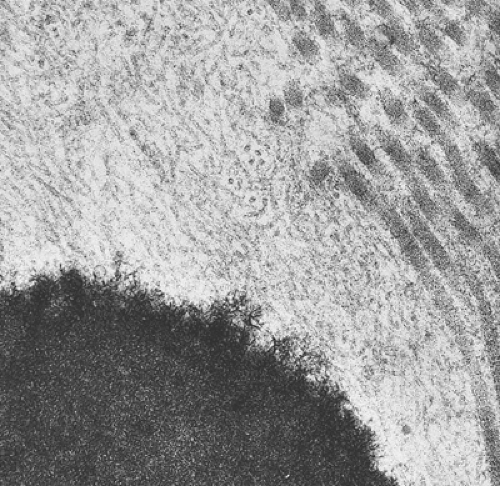 Figure 4-31 Electron microscopy of fibrous dysplasia. Mature cross-striated collagen fibrils are seen (upper right corner) as well as an extended area with immature collagen fibrils. Mineralization of the matrix progresses (lower left) regardless of the structure and maturation of the collagen. |
Polyostotic Fibrous Dysplasia
Clinical Presentation
Although radiographically similar to the monostotic form, polyostotic fibrous dysplasia often exhibits a somewhat more aggressive appearance. Moreover, the lesions are distributed differently in the skeleton, showing a striking predilection (more than 90% of cases) for one side of the body. They often involve the pelvis, followed in frequency by the long bones, skull, and ribs; the proximal end of the femur is a common site of occurrence (Fig. 4-32). In general, the lesions of polyostotic fibrous dysplasia progress in number and size until skeletal maturity is reached, after which they become quiescent. Only 5% of lesions continue to enlarge. Unlike the monostotic form, this variant of fibrous dysplasia is usually symptomatic. In most patients the presenting symptoms, such as a limp, leg pain, or a pathologic fracture, result from skeletal involvement. However, at a very early age, associated endocrine abnormalities (such as vaginal bleeding) usually appear first (119).
Polyostotic fibrous dysplasia that is associated with endocrine disturbances (i.e., premature sexual development, gigantism or acromegaly, hyperthyroidism, hyperparathyroidism, and Cushing syndrome) and skin pigmentation (café-au-lait spots) constitutes the disorder called McCune-Albright syndrome (59,106). This condition, which is caused by somatic mutations in the
GNAS1 gene located in chromosome 20q13 (68), appears to predominantly affect girls, who exhibit true sexual precocity resulting from accelerated gonadotropin release by the anterior lobe of the pituitary. However, a recent study indicates that, in a substantial number of cases, children treated for isolated fibrous dysplasia actually have unrecognized McCune-Albright syndrome, which becomes apparent when appropriate tests are performed (80). Typically, the café-au-lait spots of McCune-Albright syndrome show irregular, ragged (“coast of Maine”) borders, in contrast to the smoothly marginated (“coast of California”) café-au-lait markings seen in neurofibromatosis. Mazabraud syndrome is a condition in which multiple fibrous and fibromyxomatous soft tissue tumors occur in association with polyostotic fibrous dysplasia (79,89,120). In this syndrome, which was first described by German pathologist F. Henschen in 1926 (84) and later reemphasized by French physician A. Mazabraud in 1967 (105), it is important to recognize the soft tissue masses as benign myxomas (60,63,95,109) and not to confuse them with malignant soft tissue tumors that may develop de novo (e.g., MFH, malignant mesenchymoma, or liposarcoma), or those that may be present in cases of malignant transformation of fibrous dysplasia.
GNAS1 gene located in chromosome 20q13 (68), appears to predominantly affect girls, who exhibit true sexual precocity resulting from accelerated gonadotropin release by the anterior lobe of the pituitary. However, a recent study indicates that, in a substantial number of cases, children treated for isolated fibrous dysplasia actually have unrecognized McCune-Albright syndrome, which becomes apparent when appropriate tests are performed (80). Typically, the café-au-lait spots of McCune-Albright syndrome show irregular, ragged (“coast of Maine”) borders, in contrast to the smoothly marginated (“coast of California”) café-au-lait markings seen in neurofibromatosis. Mazabraud syndrome is a condition in which multiple fibrous and fibromyxomatous soft tissue tumors occur in association with polyostotic fibrous dysplasia (79,89,120). In this syndrome, which was first described by German pathologist F. Henschen in 1926 (84) and later reemphasized by French physician A. Mazabraud in 1967 (105), it is important to recognize the soft tissue masses as benign myxomas (60,63,95,109) and not to confuse them with malignant soft tissue tumors that may develop de novo (e.g., MFH, malignant mesenchymoma, or liposarcoma), or those that may be present in cases of malignant transformation of fibrous dysplasia.
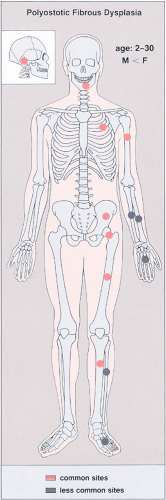 Figure 4-32 Polyostotic fibrous dysplasia: skeletal sites of predilection, peak age range, and male-to-female ratio. |
The most common complication of polyostotic fibrous dysplasia is a pathologic fracture that, when it occurs in the femoral neck, frequently leads to a “shepherd’s crook” deformity (Fig. 4-33). Occasionally, accelerated growth of a bone or hypertrophy of a digit may be observed. The development of a sarcoma in fibrous dysplasia is extremely rare, but it may occur either spontaneously (93,114,116,123) or, more commonly, after radiation therapy (115,123).
Imaging
The characteristic radiographic changes of polyostotic fibrous dysplasia may occur in any portion of the long bones, and involvement of the bone by the lesion may be limited or extensive. The lower extremities are most commonly affected, with a preferentially unilateral distribution. As in the monostotic form, the articular ends of bones are usually unaffected. This latter phenomenon has given rise to speculations that a defect of the primary ossification center may lead to impairment of the production and maintenance of mature cancellous bone. The cortex, as in the solitary form, is usually intact but is often thinned because of the expansive nature of the lesion. The borders of the lesion are well defined, and the inner cortical margins may exhibit scalloping. The variable amount of bone in the fibrous tissue that replaces the cancellous bone imparts a radiographic picture that ranges from lucency to increased density (Fig. 4-34A). More fibrous lesions exhibit greater lucency, occasionally with a loss of the trabecular pattern, and a ground-glass, milky, or smoky appearance (Fig. 4-34B). Massive formation of cartilage may be observed in the lesion, accompanied by secondary calcification and ossification patterns, known as fibrocartilaginous dysplasia or fibrochondrodysplasia (76,85,88,97), that can easily be confused radiographically with a cartilaginous neoplasm (Fig. 4-35) (72,74). This condition should not be confused with so-called focal fibrocartilaginous dysplasia of long bones (66), which occurs mainly in children and young adults. Characteristically, it affects the proximal tibia, although other long bones, such as the ulna and femur, may sometimes be involved. Radiographic examination ordinarily shows a radiolucent lesion in the medial metadiaphyseal segment of the bone and perilesional cortical thickening (Fig. 4-36).
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


