Fig 6.1
Location of ATM gene on long arm of chromosome 11
ATM gene is, bidirectionally, transcribed and there is an intergenic region spanning 520 bps within this gene which is shared by the flanking gene named NPAT. The NPAT protein is induced by E-Cdk2 kinase to activate the expression of histone proteins during G1-S transition and early S phases (Gao et al. 2003).
There is a long list of ATM mutations containing approximately 400 alterations that most of them (70 %) lead to production of truncated protein and are 100 % penetrant (Concannon and Gatti 1997). Missense alterations have usually lower penetrance and are associated with milder phenotypes. Of note, AT patients are typically heterozygous for more than one mutation (compound heterozygous) and homozygous patients were hardly detected. No hotspot region was identified in ATM gene indicating that most of the ATM mutations are unique (Gatti et al. 1999), however this matter is diverse in different populations.
The ATM gene is predominantly expressed in adipose, blood, brain, adrenal, breast, colon and heart tissues.
6.1.3 ATM Protein
ATM protein encoded by ATM gene, harbors 3056 amino acids and weights 350 kDa. It is a member of a large serine tyrosine kinase family called Phosphatidylinositol 3-kinase-related kinases (PIKKs) . The name of this superfamily is due to the presence of a similar sequence in its members and phosphatidylinositol 3-kinases (PI3Ks) . The other members of this family include ATM- and RAD3-related (ATR), DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and mammalian target of Rapamycin (m-TOR) (Wyman 1998).
There are three known isoforms of ATM protein. The first isoform constitutes of 3056 amino acids and weights for 350.6 kDa which has shown high degree of homology with mammalian, Drosophila and yeast PI3 kinase. The second and the third isoforms have 1708 and 131 amino acids, respectively. The mouse ATM is more close to the isoform 1 which contains 3066 amino acids and is 84 % similar to human ATM in sequence (Pecker et al. 1996).
There are five major domains within the structure of ATM protein with different characteristic functions (Fig. 6.2). These domains include Huntington, Elongation factor 1A, protein phosphatase 2A A-subunit, TOR (HEAT) repeat domain . The HEAT domain lies at the N-terminal of ATM protein and it has several HEAT repeat of amino acids. HEAT stands for histidine (H), Glutamic acid (E), Argenine (A) and tyrosine (T) amino acids. It makes connection between ATM and NBS1, P53, c-Abl proteins to be involved in repair of DNA double strand break (DDB) (Perry and Kleckner 2003). The FRAP-ATM-TRRAP (FAT) and kinase (KD) domains follow the HEAT domain and interact together to stabilize the C-terminal of ATM protein. KD performs the kinase activity of ATM protein which is modulated by the adjacent domains of ATM protein including PIKK-regulatory domain (PRD) and the FAT-C-terminal (FATC) domain . The FATC domain may be the most conserved domain of ATM protein and is composed of 30 amino acids which form α-helix structure through disulfide bonding (Savitsky et al. 1995b). It was shown that FAT domain has also α-helix structure which in its tertiary structure provides a tunnel to enable the ATM to wrap around the damaged DNA. The importance of translational modification of FAT and FATC domains in regulation of ATM activity shows that they play pivotal role in modulating the conformational changes in ATM structure defining its activity. The acetylation of C2991 site within the FATC domain determines the phosphorylation status of S1981 site of FAT domain which is critical for gathering the non-functional dimers together (Fig. 6.2) (Rivera-Calzada et al. 2005).
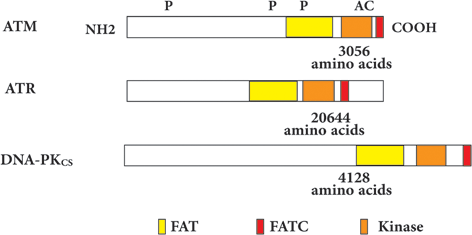
Fig. 6.2
Shared domains in PIKKs family members and the sites of post-translational modifications. (Modified from McKinnon 2012)
Post-translational modifications of ATM almost occur in response to DNA damage and include phosphorylation at various sites of protein and acetylation. One of the most important of these modifications is phosphorylation at serine 1981 which was recently found to be necessary for ATM signaling and its activation (Bakkenist and Kastan 2003). It was stated that there was no effect on the overall process of DNA repair in mice whom were knocked out for other modifications, however, their critical roles in ATM signaling remains to be well elucidated (Daniel et al. 2008; So et al. 2009).In addition, the ATM protein status doesn’t change in location and amount during all of the phases of cell cycle (Brown et al. 1997).
6.2 ATM Functions
ATM is well known for its critical role as a core of a triangle including DNA repair, cell cycle control and apoptosis. Therefore, the major functions of ATM can be descried for each apex of the mentioned triangle in details which are correlated to each other. The fate of ATM signaling depends on the determining factors including type of tissue and cell in addition to the stage of development wherein DNA damage has been occurred.
6.2.1 DNA Repair
ATM is well known for its critical activity in response to damage-specific DNA binding (DDB) protein to activate the main cellular check points avoiding the replication of damaged DNA and leading it to senescence or apoptosis. However, there are some evidences relying on the role of ATM in various other pathways induced by oxidative stress as a consequence of metabolic aberrations including autophagy (Zhang et al. 1997).
DDB is the most deleterious damage which could be occurred at DNA level. Various exogenous and endogenous factors are responsible for creating DDB . The first includes ionizing radiation not UV light and the latter comprises of toxic elements such as free radicals produced in oxidative metabolism. In addition, mitotic recombination during T cell and immunoglobulin genes rearrangement is also recognized as one of the most important cause of DDB (Wyman and Kanaar 2006).
It is interesting to note that ATM is actively involved in 10 % of DDB which affects the heterochromatin DNA (Goodarzi et al. 2008). In this way, two proteins including 53 BP1 and KRAB associated protein 1 (KAP-1) are essential through which phosphorylation of the latter by ATM leads to alter the conformation of heterochromatin structure required for initiation of DNA repair. The 53BP1 enables ATM to damage site, activate KAP-1 protein and inducing repair in the late breaking DNA (Goodarzi et al. 2008; Ziv et al. 2006; Noon et al. 2010).
DDB can be repaired through two alternative pathways including homologous recombination (HR) and non-homologous end-joining (NHEJ). When DNA is targeted to be repaired by NHEJ pathway, the two ends of damaged DNA could be detected and occupied by highly conserved Ku70–Ku80 kinase heterodimer recruiting Artemis nuclease; these events are followed by an inaccurate cutting process of the broken DNA and leave the primary site for polymerization activities of µ and λ DNA polymerases. In yeast, when no homologous sequence being found to be as template of polymerization, NHEJ is driven through recognizing the broken ends by DNA–PKcs which interacts with Ku heterodimer. DNA–PKcs is another important member of PIKKs family that has a pivotal role in checkpoint signaling (Gottlieb and Jackson 1993; Dvir et al. 1992). Subsequently, the newly synthesized DNA segment being ligated by XRCC4 accompanying DNA ligase IV in a ligation complex without prior processing of the DNA ends. Of note, the homologous of DNA ligase IV, Cernunnos, and XLF factor encourage the initiation of ligation process in all animal species (Buck et al. 2006; Ahnesorg et al. 2006; Cavero et al. 2007). It is interesting to note that DNA–PKcs brings two ends of both strands of broken DNA together. It is alternatively carried out by Mre11–Rad50–Xrs2 (MRX) complex or other proteins in yeast (Manolis et al. 2001).
Although it is highly proficient, imprecise cutting and replacing the DNA nucleotides is the major reason for calling NHEJ as an error and mutation prone pathway of DNA repair (Pitcher et al. 2005; Lees-Miller and Meek 2003; Lieber 2008). NHEJ is the choice of DNA repair when the damage takes place in G0, G1 and in the initial times of S or replication phases.
Another choice of DDB repair is HR in which the broken part of DNA is replaced based on the sequence of sister chromatid as template of polymerization. This turn, the call for initiation of DNA repair is arisen when the ends of broken double strand DNA is filled with a triple protein complex known as MRN complex (MRE11–RAD50–NBS1/Xrs2). After cutting both flanking sites of broken part of DNA, replicated protein A or RPA binds to the created single strands and prevents rejoining of them until the polymerization of their corresponding opposite strands occur (Kadyk and Hartwell 1992; Sonoda et al. 2001). In yeast, RPA is dislodged from the single stranded DNA by cooperation between Rad51 and Rad52 proteins to provide the condition for initiation of polymerization. Rad51 actually carries out the task of search for the homologous sequence to be as template for replacing the missing cut nucleotides through wrapping around the nucleosome filaments (Sung 1994; Ogawa et al. 1993).
HR is the preferred choice of post replication DNA repair within the late S phase and throughout the G2 phase. The role of ATM in HR pathway may be tissue specific as testis and usually includes phosphorylation and activation of less critical elements of HR pathway (Barlow et al. 1997). In spite of the fact that HR is free of any mistake or mutation in replacing the missing broken DNA double strand sequences, it is intriguing to say that the cell prefers to use the error prone NHEJ pathway. Although the recent studies have refuted it, it may be due to the tight and complex infrastructure of DNA sequence wrapping around the histone proteins making homology searching more impossible and difficult (Ciccia and Elledge 2010; Bensimon et al. 2011; Kim et al. 1999; Matsuoka et al. 2007). However, the choice of DNA repair pathway is phase specific and it seems that it is the matter of authority, so cell determines and selects the appropriate one according to the cellular phase in which break occurs.
There are many reports on diseases including cancer and neurological disorders which are as consequences of defects in elements of either HR or NHEJ DNA repair pathways (Mu et al. 2007; Bensimon et al. 2010; Bennetzen et al. 2010; Marzano et al. 2012; McKinnon 2012; Schalch et al. 1970).
When DDB occurs, ATM is recruited to damage site through formation of MRN complex at the end of broken DNA (Shiloh 2003). Of note, Mre11 acts as an endonuclease, resects the broken DNA segment which should be replaced. Interaction of Nbs1 with C-terminal binding protein interacting protein (CtIP) is another fact indicating that MRN complex is actively contribute to DDB repair. In addition, attachment of MRN complex to damaged DNA leads to extension of coiled coil structure of Rad50 which makes the connection possible between two MRN complexes residing on both opposite DNA strands (Wiltzius et al. 2005; Hopfner et al. 2002).
It is assumed that recruitment of MRN complex to damage site is triggered by the presence of highly conserved identified motifs in Nbs1 whereas motifs in KU and ATR interacting protein (ATRIP) are required for motivating the recruitment of DNA-PKcs and ATR to damage site, respectively (Falck et al. 2005). ATM is tightened to the damage site through interaction of its C-terminus to Nbs1 protein. The same condition is held for ATRIP/ATR and KU/DNA-PKcs (Williams et al. 2009). However, some other proteins have been identified to be involved in activation of ATM after DDB including checkpoint protein, ring finger protein 8 and E3 ubiquitin ligases (Wu et al. 2011).
Dynamic ATM activation is dependent on the phosphorylation of S1981 within the FAT domain releasing the inactive ATM dimer to be bound to other molecules (Bakkenist and Kastan 2003). Phosphorylation and dephosphorylation of S1981 is performed by PP2A and WIP1 phosphatase, respectively (Shreeram et al. 2006). The same site of phosphorylation is targeted by ATM on tyrosyl phosphodiesterase 1(TDP1) which plays important role in DDB repair (Das et al. 2009). It was found that WIP1 regulates the methylation status of DNA sequences through interaction with ATM/BRCA1 to recruit heterochromatin protein 1 (HP1) and subsequently DNA methyltransferases (Filipponi et al. 2013). Moreover, there are other important serines as well as Ser367, Ser1893 and Ser2996 that their autophosphorylation have revealed major impacts on ATM signaling (Kozlov et al. 2011). Besides PP2A and WIP1, PP5 are another phosphatase which modulates the phosphorylation of ATM (Goodarzi et al. 2004).
Although the critical effect of ATM autophosphorylation on DDB repair have been confirmed in human cell line studies, animal and in vitro assays couldn’t identify its importance. Activation of monomer ATM, in turn drives a cascade of phosphorylation and ubiquitinilation of several downstream molecules ending in DNA repair or apoptosis. There are some mediatory proteins which enhance and facilitate the relay of signals to target elements within the pathway (Li and Zou 2005; McGowan and Russell 2004).
The immediate downstream targets of ATM and ATR are Chk2 and Chk1 proteins, respectively.
However, when both ATM and ATR become activated, formation of a complex is initially necessary for Chk1 and Chk2 stimulation. This complex comprises of four BRCT domain containing proteins including BRCA1, MDC1/NFBD1, MCPH1/BIRT1 that taken together recruit all the critical factors involved in cell cycle checkpoint and DNA repair.
BRCA1 is phosphorylated by both ATM and Chk1 in response to DNA damage and resides on damage site. It has critical role in two intra-S and G2-M checkpoints (Deng 2006; Ouchi 2006).
The 53BP1 is required for p53 stabilization and is also involved in Chk2 phosphorylation and triggering intra-S and G2-M checkpoints when the DNA damage is occurred. There are approximately 15 sites within this protein which are targeted for phosphorylation by ATM and ATR in response to UV radiation (Wang et al. 2002).
Knock out study of MDC1 (mediator of DNA damage checkpoint protein 1) has led to disability of cell to induce its cycle checkpoints and apoptosis (Stewart et al. 2003).
In addition to recruitment of DNA repair complex, MCPH1 marks damaged DNA, which is wrapped in chromatin structure, open through interaction with SW1/SNF to be available for repair proteins (Peng et al. 2009). However, it will be discussed in further details in Chap. 7.
It is accepted that ATM and its counterpart molecule, ATR recognize and phosphorylate the serine and threonine within the S/T-Q motif of their substrates. It was determined that ATM and ATR recognize more than 900 S/T-Q motifs within about 700 target proteins (Shiloh 2003). ATM, initially, phosphorylates c-Abl which itself phosphorylate and activates Rad51, Rad52 and H2AX molecules through its special kinase function. Moreover, c-Abl phosphorylation is required for autophosphorylation of ATM at S1981 to help it to be maintained active. Phosphorylated H2AX or γ-H2AX (phosphorylation on SQE motif) marks the damaged DNA to be repaired through recruiting proteins responsible for the relevant pathway (Wang et al. 2011).
6.2.2 Cell Cycle Control
Most of the ATM functions in both cell cycle control and DNA repair pathways are overlapped and linked together. However, after the firing the phosphorylation cascade of DDB ’s repair, ATM switches its role from DNA repair to cell cycle control. It was shown that cells lacking functional ATM have aberrant G1-S, intra S and G2-M cell cycle checkpoints (Barlow et al. 1996; Borghesani et al. 2000; Elson et al. 1996; Herzog et al. 1998).
Study on postmitotic neurons has demonstrated that it is initiated through phosphorylation of ATM by cyclin-dependent kinase 5 (Cdk5) at S794. This phosphorylation is necessary for the next one on S1981 ensuring intact ATM signaling. The immediate targets of ATM phosphorylation are Chk1 and Chk2 which in turn phosphorylate p53 and Cdc25. The p53 phosphorylation stabilizes it to drive DNA damage response and when the Cdc25 become phosphorylated it will be no longer able to activate CycA- and CycE-Cdk2, leading to cell cycle stalling at G1/S transition (Lukas et al. 2003). As described above, H2AX is one of the major substrates of ATM which has shown that it shares common functions with ATM in DNA repair when damage has been occurred in G1 phase. However, it was found that H2AX can, independently, operate ATM in repairing DNA after its replication (Furuta et al. 2003).
ATM is also affects the cell cycle in S or DNA replication phase which is fulfilled through activation of BRCA1 associated surveillance complex (BASC) leading to induction of p21 followed by either apoptosis or cell cycle arrest (Fig. 6.3).
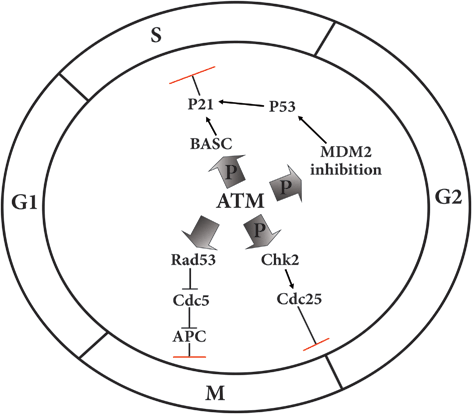
Fig. 6.3
Regulatory effects of ATM on the major cell cycle checkpoints
Activation of CycB-Cdk1 CycB-Cdk1 also named as metaphase promoting factor (MPF) by Cdc25C is necessary for transition of cell cycle from G2 to metaphase. When DNA has been damaged, Wee1 protein provides a delay in S-G2 transition and by phosphorylation and inactivation of MPF arrests the cell cycle to enter the mitosis phase. In fission yeast, there is another kinase protein called Mik1 which function besides Wee1 to stall the cell cycle before getting entry to mitosis. In addition, phosphorylation of Cdc25 by ATM induced Chk2, would be another reason that the MPF being inactivated.
Finally, the other major contribution of ATM in cell cycle control is fulfilled through phosphorylation followed by inactivation of MDM2 setting p53 free to induce the expression of p21 gene. The p21 protein is a putative suppressor of the entire cyclin-Cdk complex thought the cell cycle, so the cycle will be stopped to be repaired (Bartek and Lukas 2001).
Mitosis phase control of an ATM analogue known as Mec1 is performed by its activation after DNA damage leading to induction of Chk1 and Rad53 (Chk2 homologouse in yeast). Rad53 in turn, suppresses the Cdc5 which is the major factor of exiting from anaphase through induction of APC and destruction of Cdk1-Cyclin B. Therefore, Mec1 hinders the anaphase transition and controls the cell cycle at this main checkpoint when the DNA has been damaged (Zhang et al. 2009) (Fig. 6.3).
6.3 Other Functions of ATM Kinase
6.3.1 ATM and Telomere Length
It is worth to note that the role of ATM in telomere integrity and length is more important and critical than DNA repair in yeast. The similarity in structure of yeast proteins involved in telomere length (TL) including TEL1 and rad3 and their human homologous ATM could support the correlation between ATM and TL (Pandita and Dhar 2000). The presence of various chromosomal abnormalities as well as chromosomal breakage and short TL in AT patients could be the further evidences. It was shown that the putative contribution of ATM in DDB repair is not conserved in yeast (Peng et al. 2009).
Treatment of isolated cells either somatic or germ cells derived from AT patients have demonstrated that interaction between nuclear matrix and telomere required for telomere and chromosomal stability is governed by ATM (Pandita and Dhar 2000). ATM transfection of fibroblast cells improved the described interaction while decreased the overall rate of chromosomal breakage. Directed mutagenesis of ATM in neuroblast cells of Drosophila have shown that it is essential for maintaining the chromosomal stability and integrity and avoids of end to end chromosome fusion (Silva et al. 2004). Further studies are merited to focus on why only specific cells such as neurons are more sensitive to ATM deficiency producing neurologic symptoms in AT patients.
It was demonstrated that ATM and Mre11 are recruited to telomeric foci and detect it as a DSB when the human cells committed to enter the senescence phase. It was confirmed through knocking out of ATM which was associated with the gaining the cellular capability to continue the cell cycle. Strikingly, it is assumed that there is another switch in ATM functions from telomere protection to cell cycle arrest when the cell goes to be turned off. Recently, it was revealed that ATM differentially functions in repair of interstitial and telomeric DSBs. Any DSB near the telomere remains unrepaired in AT patients due to the lack of ATM function in repair machinery. ATM deficiency was associated with higher incidence of large deletions in telomeric regions whereas the rate of small deletions and NHEJ was greatly increased. These findings propose that ATM fulfill the telomere protection rather than telomere resection during DSB repair. Absence of ATM, therefore leads to induction of NHEJ alternative repair pathway whereas its presence impedes DNA repair through protection of telomere sequences (Muraki et al. 2013). Another study on cells lacking ATM demonstrated that ATM deficiency is responsible for telomere shortening in older mice increasing the risk of cancer at higher ages (Vaziri et al. 1997).
Contribution of ATM to TL integrity was further verified that ATM deficiency makes telomeric repeat sequences untraceable to be recognized by telomerase and other proteins involved in maintaining telomere integrity (Khanna 2000).
6.3.2 ATM in Cytoplasm
The presence of some clinical presentations in AT patients which seems to be unrelated to defect in DNA repair could be strong evidence on the role of ATM in other cellular functions rather than cell cycle and DNA repair control. Although it was shown that ATM is more present in nucleus versus cytoplasm in different cell types, the distribution of it is approximately equal between nucleus and cytoplasm in neural cells of cerebellum (Yang et al. 2011; Barlow et al. 1999; Boehrs et al. 2007). Finding ATM in cytoplasm is indicative of its special functions rather than nuclear activities (Li et al. 2009). It was demonstrated that ATM was associated with cytoplasm organelles including peroxisomes and endosomes (Watters et al. 1999).
The over-expression of ATM gene upon insulin treatment and the presence of insulin resistant type 2 diabetes in AT patients are complementary findings on the critical role of ATM in insulin metabolism pathway. Actually, ATM is contributed to insulin signaling through phosphorylation of eIF-4E-binding protein 1 (4E-BP1) at serine 111 and its separation from eIF-4E translation elongation factor leading to cap dependent translation (Yang and Kastan 2000). Insulin resistance in mouse models manipulated to have no functional ATM, was a further evidence of its role in insulin metabolism (Wu et al. 2010). It is conceivable that ATM plays pivotal role in insuline induced protein synthesis and glucose metabolism and may relay the surviving signals from IGF receptor within the neural cells (Yang and Kastan 2000).
Moreover, ATM is relocated to cytoplasm within the ATM-NEMO complex when its signaling has led to cell apoptosis. NEMO or NF-κB essential modulator is phosphorylated by ATM to be transported accompanying it to cytoplasm wherein NEMO and IκB kinase interaction begins the NF-κB induced apoptosis pathway (Wu et al. 2006).
Along these lines, ATM modulates the mTOR complex 1 (mTORC1) signaling in cytoplasm through stimulation of tuberous sclerosis complex 2, LKB1 and MAPK or phosphorylation of hypoxia induced factor (HIF) to prevent autophagy (Shiloh 2003; Zoncu et al. 2010). Inability of ATM deficient cells to up regulate the HIF level may be an important evidence on this fact that it is the major sensor of cellular oxygen level (Mongiardi et al. 2011). In addition, ATM indirectly controls the mitochondrial function by modulating HIF and its complex which is consisted of HIF-1α and HIF-1β (Cam et al. 2010). The ATM interaction with HIF factor would be a good reason to propose that ATM is an active member of cancer cell metastasis pathway through inducing angiogenesis.
Involvement of ATM in mitochondrial diseases is relying on its important roles in mitochondrial functions (Ambrose et al. 2007). Given the deregulation of mitochondrial homeostasis in structure, membrane potential and respiratory activity in cells lacking ATM and ATM gene alterations in various mitochondrial diseases, it may be the guardian of mitochondrial genome in addition to nuclear genome (Inomata et al. 2009). Moreover, ATM is involved in mitochondrial biogenesis through activation of AMPK in response to ATP deficiency and indirectly avoids lipid, carbohydrate and protein synthesis pathways (Reznick and Shulman 2006). In addition, it is assumed that ATM controls the number of mitochondria, although it has not been elucidated that this control is performed in negative or positive manner. For instance In vivo, shortage of ATM in thymocytes was associated with increased number of abnormal mitochondria that their degradation led to enhanced oxidative stress described below (Valentin-Vega et al. 2012).
Strikingly, most of the downstream molecules of ATM as well as p53 or CREB finally destined to mitochondria (Ditch and Paull 2012). The critical described functions in response to oxidative stress and its byproducts would be another linkage between ATM and mitochondria. Oxidative stress means exceeding of either reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) or superoxide anion radical (●O−2) or nitrogen reactive species (RNS) over anti-oxidant reagents within the cell (Barzilai et al. 2002). It is the prominent feature of AT patients considered as a major cause of neurodegeneration in them (Ditch and Paull 2012). Of note, it was shown that the mitochondrial level of cytochrome C had been decreased in AT patients indicating the possible effect and interaction of ATM with the elements of respiratory circle (Patel et al. 2011). It was also demonstrated that ATM increases the antioxidant levels through controlling the mitochondrial pentose phosphate pathway (PPP) . Extra free electrons are indirectly neutralized by ATM through induction of glucose-6-phosphate dehydrogenase (G6PD) activity and increase in cellular levels of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) . In addition, two ATM molecules form dimer with each other through disulfide bonding between their cysteine amino acids in C2991 site. This dimer formation is necessary to overcome the inhibitory effect of ROS on DSB repair (Guo et al. 2010).
6.4 ATM Defects in Human Malignant and Non-Malignant Diseases
6.4.1 ATM and Various Cancers Predisposition
There are many evidences relying on the importance of ATM gene aberrations in development of different cancers and support its critical role as a tumor suppressor gene. Firstly, it is based on the increased risk of being affected with leukemia and lymphoma (20 %) in AT patients and secondly, decrease in ATM expression which was observed in various types of animal and human cancers (Gumy-Pause et al. 2004; Matei et al. 2006). According to the later evidence, even heterozygote carriers of ATM mutations especially amongst relatives of AT patients are prone to develop malignancies (Swift et al. 1987). It was stated that around 1 % of general populations are heterozygote for ATM gene alterations, each ATM mutation would be the focus of more assessment (Matei et al. 2006).
6.4.2 ATM Aberrations in Breast Cancer
A key question is that how much does ATM alteration raise the risk of breast cancer in AT patients, their relatives and carriers of ATM mutations? Unfortunately the precise answer is still unclear among various populations. Swift was the first one who by studying on 110 AT families, suggested the enhanced probability of breast cancer in female and male relatives of AT patients which was revealed to be 3.1 and 2.3, respectively by 1987 (Swift et al. 1987). Review on the subsequent studies on the risk of breast cancer in AT patient’s relative demonstrated that they had 3.9 % risk compared to general population (Easton 1994). Taken together with other comprehensive carried out studies, the overall risk of breast cancer is almost 2.23 more times in relatives of AT patients versus general population which is doubled in individuals who are younger than 50 years (Thompson et al. 2005).
The initial study on comparing breast cancer patients with healthy controls could not support the hypothesis of higher frequency of breast cancer incidence in carriers of ATM mutations (FitzGerald et al. 1997). It’s may be due to the different pattern of ATM mutations in AT patients and healthy carriers. Except of dominant negative and some of the splice site alterations, missense mutations usually have less effects on protein and are associated with residual function of protein versus truncating mutations influencing cancer susceptibility (Gatti et al. 1999). It was proposed that the role of ATM variants especially non-synonymous ones is not important in developing breast cancer in the carriers (Renwick et al. 2006). The ATM mutations causing AT was considered as low penetrant breast cancer alterations which are associated with two fold increase in risk of breast cancer in the carriers (Thompson et al. 2005). Moreover, the risk of breast cancer in carriers of ATM mutations was deduced to be similar to the risk of breast cancer in carriers of CHEK2*1100delC alteration (Nevanlinna and Bartek 2006).
The presence of premature stop codon mutations in ATM gene was associated with nine fold increase in risk of familial breast cancer amongst Dutch population (Broeks et al. 2000b). However, the chance of breast cancer development has shown to be two times more in British patients who were carrying ATM alterations (Inskip et al. 1999; Thompson et al. 2005). Although several studies couldn’t show the contribution of AT in pathogenesis of breast cancer, Renwick and his colleagues have demonstrated that carriers of these mutations have two fold chance to be affected by breast cancer (Renwick et al. 2006; Teraoka et al. 2001; Sommer et al. 2003).
However, there are many reports relying on the significant characters while unknown roles are assumed to be due to some nucleotide substitutions of ATM gene in development of breast cancer. The c.7271T>G alteration enhances the risk of breast cancer in either homo- or heterozygous mutated ATM individuals (Stankovic et al. 1998).
The T7271G alteration is considered as another ATM gene alteration conferring special risk of breast cancer which was identified in three AT families and one case control study. It was identified in a family in which the heterozygous mother and daughters for T7271G were affected by breast cancer in 82, 44 and 50 years old, respectively. Breast cancer was also diagnosed in three paternal aunts of two AT brothers carrying T7271G accompanying a truncating mutation of ATM gene. Five breast cancer patients were also found to be carrier of T7271G substitution in an Australian family (Stewart et al. 2001; Stankovic et al. 1998). In another study, it was found that this mutation was associated with more than 15 fold increases in risk of breast cancer with 60 % penetrance (Chenevix-Trench et al. 2002). However, further studies are warranted to elucidate the role of this substitution in pathogenesis of breast cancer.
The significance of IVS 10-6T >G splice site mutation which is associated with the loss of exon 11 of ATM gene, in susceptibility, to breast cancer has been evaluated by three different studies. Broeks and colleagues have found that carriers of this alteration are at risk for development of bilateral and early onset breast cancer (Broeks et al. 2003). It was also demonstrated that this mutation was penetrant in 17.2 % by the age of 70 years old (Thompson et al. 2005).
It is stated that being carrier for ATM gene mutations didn’t cause over-reaction to radiotherapy in breast cancer patients meriting further studies to clarify it (Broeks et al. 2000). However, whether it is safer to consider radiotherapy as an alternative option in treatment schedule of breast cancer patients carrying ATM alterations or not, that is a crucial challenge which varies between different populations.
Sporadic breast cancer is as a result of loss of heterozygousity (LOH) of various genes in 40 % of cases as the LOH of ATM is contributed in the earlier phases of breast cancer pathogenesis (Rio et al. 1998; Hampton et al. 1994). Although, LOH of ATM gene may be associated with higher risk of cancer, it is a further support for two-hit model of tumorgenesis. However, we have preliminary demonstrated the three hit hypothesis in astrocytoma which will be described in detail later in section of brain tumors.
The importance of D1853N in breast cancer was previously published. We have investigated 129 Iranian patients affected with breast cancer to determine the presence of D1853N polymorphism in patients compared to 248 healthy controls through Mutant allele-specific PCR amplification (MASA) assay confirmed by sequencing. This polymorphism was genotyped in 31.0, 26.9 and 12.5 % of the cases, internal- and external- controls, respectively. The relative risk was calculated to be 2.5. It was concluded that the significant difference between patient carriers and controls would be helpful in screening of cancer prone families (Mehdipour et al. 2011a).
6.4.3 ATM and Risk of Leukemia
ATM mutations are associated with both types of leukemia either in adult or childhood lymphoma. However, in T-cell prolymphocytic leukemia (T-PLL) patients whom are usually diagnosed in later ages, both of ATM alleles were non-functional. Actually, the LOH of ATM gene was found to be completed with a missense mutation frequently occurs in PI-3K domain encoding part of other allele (Vorechovsky et al. 1997; Stilgenbauer et al. 1997; Stoppa-Lyonnet et al. 2000). Although the LOH of ATM gene was also seen in acute lymphoblastic leukemia (ALL), it seems that it is infrequent in this type of leukemia (Haidar et al. 2000; Gronbaek et al. 2002).
Follicular center cell lymphoma (FCL) is low grade B-cell non-Hodgkin’s lymphomas (BNHL) with mild presentations (Cuneo et al. 2000) in which heterozygous deletion and missense point mutation of ATM gene were described in 11 and 8 % of all the FCL respectively.
The role of ATM gene alterations in pathogenesis of B-cell chronic lymphocytic leukemia (B-CLL) sounds to be more important as they were demonstrated in 26–40 % of this type of leukemia (Bullrich et al. 1999; Starostik et al. 1998; Stankovic et al. 2002; Schaffner et al. 1999). Missense mutations were found to be the leading type of ATM mutations in B-CLL patients which are usually associated with lower expression of ATM protein in homozygous status (Starostik et al. 1998). The B-CLL patients carrying ATM mutations had shown more severe phenotype than mutation in other genes that it may be due to their disability to take care of DNA against damages induced by treatment or every endogenous stresses (Stankovic et al. 2004). It was confirmed in another study on 57 CLL patients that, ATM gene was deleted or had point mutation in 25 % of patients (Guarini et al. 2012).
The t(11; 14) (q13;q32) translocation and 11q deletion are the frequent ATM alterations which were found in Mantle cell lymphoma (MCL) . A microarray analysis on all types of lymphoma had revealed that MCL owned the highest frequency of ATM alterations, even though no association was found between ATM mutations and clinical presentations (Fang et al. 2003).
Diffuse large B-cell lymphoma (DLBCL) is the other type of lymphoma in which ATM mutations were determined in 13–20 % of these patients. However, no evidence was found relying on the hypermethylation of ATM promoter in DLBCL patients (Fang et al. 2003; Gronbaek et al. 2002).
Germ line ATM alterations were almost detected in Hodgkin’s diseases (HD) that have increased the risk of breast cancer after radiotherapy (Broeks et al. 2000a; Offit et al. 2002).
Although there are a few reports on the role of ATM mutations in childhood leukemia, it was demonstrated that ATM alterations were found in 25 % of ALL patients which was significantly associated with the disease relapse (Gumy Pause et al. 2003).
6.4.4 ATM Role in Brain Tumors
Barlow and colleagues initially found that ATM protein has cytoplasmic localization in Purkinje cells as well as cerebellum. They demonstrated that the presence of ATM protein in cytoplasm is necessary and critical for balancing the number of cytoplasmic organelles, in particular lysosome. Accumulation of lysosomes in cytoplasm found in ATM−/− knocked out mice and was not associated with any neuronal degeneration (Barlow et al. 2000). Since then, ATM has been the focus of another study to determine the correlation between its expression and radioresistancy in glioblastoma multiform (GBM) in multiple GBM cell lines. Although there was inconsistent pattern in ATM expression among various types of cell, it was described that reducing the ATM expression may help to alleviate the mode of radio-resistance in GBM patients (Tribius et al. 2001).
Investigation of ATM gene mutations was followed in one GBM cell line (M059J) through yeast-based frameshift/stop codon assay. It was found that the absence of catalytic subunit of DNA-PKcs beside a truncating mutation in ATM were responsible for hypersensitivity of M059J to radiation (Tsuchida et al. 2002). However, in the attempts to find ATM mutations in medulloblastomas, no success was achieved in tumor samples, while the LOH of 11q region was identified in 25 % of the patients. Of note, the two putative D1853N and F858L polymorphisms of ATM gene were identified in approximately 20 % of patients (Liberzon et al. 2003).
We have described the D1853N polymorphism in a patient affected with astrocytoma and proposed the three hit hypothesis (Mehdipour et al. 2008). Based on this hypothesis, first alteration was inherited through germ line and the two novel splice sites including IVS 38- 63T→A and IVS38- 30 A→G polymorphisms constitute the second and third hits. Of note, D1853N polymorphism was found in one allele whereas the other two polymorphisms were found in another allele (Mehdipour et al. 2008). D1853N polymorphism was transmitted from proband’s mother while the IVS 38- 63T→A polymorphism has been arisen in the early zygotic stage before differentiation of the peripheral blood. Although, the well-known two hits hypothesis proposed by knudson is a putative mechanism for tumor evolution, it was not shown in other cancer types rather than retinoblastoma.
Assessment of multiple loci LOH including ATM on brain metastasis in sporadic breast cancer patients demonstrated that multiple loci LOH have reduced the survival after brain metastasis (Hampl et al. 1998).
Regarding the ATM expression on the fresh brain tumor tissue samples, we have assayed the expression of mRNA of Cyclin D2, P53, Rb and ATM genes in 52 brain tumor patients by Real time polymerization chain reaction (Real time PCR) (Kheirollahi et al. 2011). It was demonstrated that ATM, Rb, P53 and Cyclin D2 had higher expression in astrocytoma compared to meningioma samples. However, ATM and Cyclin D2 genes had higher expression in higher grades of astrocytoma while declined expression was detectable in Rb and P53 genes. As a complementary insight and by considering the nature of protein expression, expression of ATM and p53 is reflective of low expression of ATM and p53 protein in tumor cells of patient affected with meningioma. In contrast both proteins revealed to have diverse pattern as two clone of cells including one with low and another clone with high expression in patient with astrocytoma. As it is expected, both protein expression was high in control (Fig. 6.4).
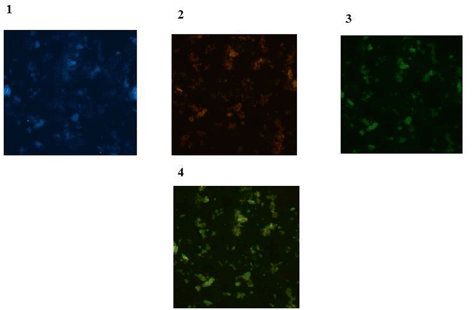
Fig. 6.4
Protein expressions of ATM and p53 in brain tumor cells. 1 Tumor cells of a patient with meningioma with dapi (blue); 2 Same cells conjugated with R-Pe (orange) representative of ATM protein characterized with low expression by IF. 3 Same cells conjugated with FITC (green), reflecting low expression of p53 protein; 4 Merged image of ATM and p53 presenting harmonized cooperation between these two proteins
However, the image of Cyclin D2/Cyclin E/C-Fos in a patient affected with astrocytoma is provided (Fig. 6.5). In this figure, there is more harmony between Cyclin D2 and c-Fos, but the status of protein expression in Cyclin E is rather low. Furthermore by considering the key role of cell cycle targets and transcription factor through tumor progression and to highlight the diverse pattern of protein expression in different pathological classifications of brain tumors, expression profile of glioblastoma multiform,

Fig. 6.5
Protein expression of Cyclin D2, Cyclin E and C-Fos in a patient affected with astrocytoma is provided . 1 Brain tissue cells of a patient affected with astrocytoma with dapi; 2 Same cells conjugated with Pe-cy5 reflecting diverse expression of Cyclin D2 including low and high; 3 The same cells conjugated with R-Pe illustrating very low expression of Cyclin E; and 4 Same cells with FITC presenting mixed expression mode including low, medium and high. In this figure, there is more harmony between Cyclin D2 and c-Fos, but the status of protein expression in Cyclin E is rather low. (From P.Mehdipour’s archive)
astrocytoma and meningioma has been provided (Fig. 6.6). In addition, expression profile of ATM/Rb/Ki67 in a meningioma patient is reflective of low expression of Ki 67, and more coordination between ATM and Rb protein (Fig. 6.7). To explore the crucial role of protein expression at cellular level, more complementary images and data is available in our recent publication (Mehdipour et al. 2014).

Fig. 6.6
 Reduced Risk of Cancer in Schizophrenia, a Bridge Toward Etio-Pathology and Therapy of Both Diseases
Reduced Risk of Cancer in Schizophrenia, a Bridge Toward Etio-Pathology and Therapy of Both Diseases
 Biodynamic Phenotypic and Epigenetics Changes of Circulating Tumor Cells: Their Application in Cancer Prognosis and Treatment
Biodynamic Phenotypic and Epigenetics Changes of Circulating Tumor Cells: Their Application in Cancer Prognosis and Treatment
 An Introduction to Impact of Bio-Resonance Technology in Genetics and Epigenetics
An Introduction to Impact of Bio-Resonance Technology in Genetics and Epigenetics
 Epigenetics of Thyroid Cancer
Epigenetics of Thyroid Cancer
 Malignant Rhabdoid Tumor: Epigenetic Mechanism of Tumorogenesis
Malignant Rhabdoid Tumor: Epigenetic Mechanism of Tumorogenesis
 Sentinel Gene Within Cell Territory and Molecular Platforms in Cancer: Methylation Diversity of p53 Gene in Brain Tumors
Sentinel Gene Within Cell Territory and Molecular Platforms in Cancer: Methylation Diversity of p53 Gene in Brain Tumors




Protein expressions of cyclin D2, CyclinE and c-Fos in brain tumor cells of patients affected with brain tumors. GBM Gleioblastoma multiform. 1 Tumor cells of brain tissue of a patient affected with GBM. a Tumor cells with dapi filter (blue), as counter stain; b cells conjugated with Pe-cy5 (Texas red) reflect overexpression of Cyclin D2; c same cells conjugated with, R-pe reflecting low expression of cyclin E protein; and d same cells conjugated with FITC (green), representative of c-Fos protein characterized with very low expression. 2 Tumor cells of brain tissue of a patient affected with astrocytoma. a Tumor cells with dapi filter (blue), as counter stain; b cells conjugated with Pe-cy5 (Texas red) reflect relativelyover-expression of Cyclin D2; c same cells conjugated with, R-pe reflecting low expression of cyclin E protein; and d same cells conjugated with FITC (green), representative of c-Fos protein characterized with very low expression. 3 Tumor cells of brain tissue of a patient affected with meningioma. a Tumor cells with dapi filter (blue), as counter stain; b cells conjugated with Pe-cy5 (Texas red) reflect both low and high expression of Cyclin D2; c same cells conjugated with, R-pe reflecting low expression of cyclin E protein; and d same cells conjugated with FITC (green), representative of c-Fos protein characterized with high expression. Magnification: x100. (Adopted from Mehdipour et al. 2014: P.Mehdipour’s Archives)
Related posts:
Reduced Risk of Cancer in Schizophrenia, a Bridge Toward Etio-Pathology and Therapy of Both Diseases
Biodynamic Phenotypic and Epigenetics Changes of Circulating Tumor Cells: Their Application in Cancer Prognosis and Treatment
An Introduction to Impact of Bio-Resonance Technology in Genetics and Epigenetics
Epigenetics of Thyroid Cancer
Malignant Rhabdoid Tumor: Epigenetic Mechanism of Tumorogenesis
Sentinel Gene Within Cell Territory and Molecular Platforms in Cancer: Methylation Diversity of p53 Gene in Brain Tumors
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
