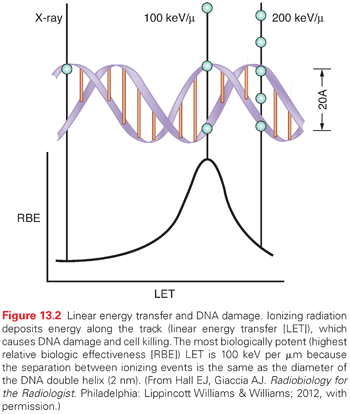
The direct effects of radiation are the consequence of the DNA in chromosomes absorbing energy that leads to ionizations. This is the major mechanism of DNA damage induced by charged nuclei (such as a carbon nucleus) and neutrons and is termed high linear energy transfer (Fig. 13.2). In contrast, the interaction of photons with other molecules, such as water, results in the production of free radicals, some of which possess a lifetime long enough to be able to diffuse to the nucleus and interact with DNA in the chromosomes. This is the major mechanism of DNA damage induced by x-rays and has been termed low linear energy transfer.1
A free radical generated through the interaction of photons with other molecules that possess an unpaired electron in their outermost shell (e.g., hydroxyl radicals) can abstract a hydrogen molecule from a macromolecule such as DNA to generate damage. Cells that have increased levels of free radical scavengers, such as glutathione, would have less DNA damage induced by x-rays, but would have similar levels of DNA damage induced by a carbon nucleus that is directly absorbed by chromosomal DNA. Furthermore, a low oxygen environment would also protect cells from x-ray–induced damage because there would be fewer radicals available to induce DNA damage in the absence of oxygen, but this environment would have little impact on DNA damage induced by carbon nuclei.2
Cellular Responses to Radiation-Induced DNA Damage
Checkpoint Pathways
The cell cycle must progress in a specific order; checkpoint genes ensure that the initiation of late events is delayed until earlier events are complete. There are three principal places in the cell cycle at which checkpoints induced by DNA damage function: the border between G1 phase and S phase, intra-S phase, and the border between G2 phase and mitosis (Fig. 13.3). Cells with an intact checkpoint function that have sustained DNA damage stop progressing through the cycle and become arrested at the next checkpoint in the cell cycle. For example, cells with damaged DNA in G1 phase avoid replicating that damage by arresting at the G1/S interface. If irradiated cells have already passed the restriction point, a position in G1 phase that is regulated by the phosphorylation of the retinoblastoma tumor suppressor gene (Rb) and its dissociation from the E2F family of transcription factors, they will transiently arrest in S phase. The G1/S and intra-S phase checkpoints inhibit the replication of damaged DNA and work in a coordinated manner with the DNA repair machinery to permit the restitution of DNA integrity, thereby increasing cell survival.
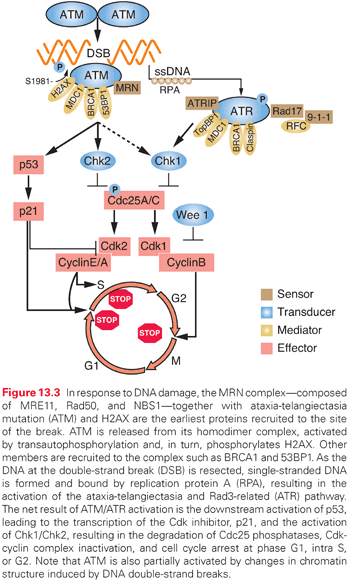
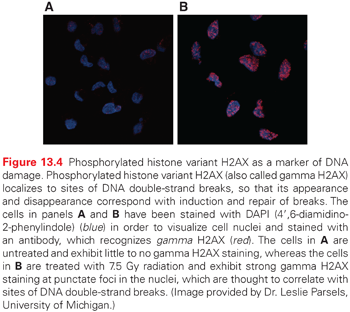
The earliest response to radiation is the activation of ataxia-telangiectasia mutated (ATM), which involves a conformational change that results in the activation of its kinase domain and phosphorylation of serine 1981 (see Fig. 13.3).3 This phosphorylation causes the ATM homodimer to dissociate into active monomers that phosphorylate a wide range of proteins such as 53BP1, the histone variant H2AX, Nbs1 (Nijmegen breakage syndrome; a member of the MRN complex, composed of Mre11, Rad50, and Nbs1), BRCA1, and SMC1 (structural maintenance of chromosomes), and these proteins coordinate repair with the cell cycle.4 In response to DNA damage, H2AX is rapidly phosphorylated by ATM and localizes to sites of DNA double-strand breaks in multiprotein complexes described as foci (Fig. 13.4). Phosphorylation of H2AX by ATM results in the direct recruitment of Mdc1 and forms a complex with H2AX to recruit additional ATM molecules, forming a positive feedback loop.
The G1/S phase checkpoint is the best understood. In response to DNA damage, activated ATM can directly phosphorylate p53 and mdm2, the ubiquitin ligase that targets p53 for degradation. These phosphorylations are important for increasing the stability of the p53 protein. In addition to ATM, checkpoint kinase 2 (Chk2) also phosphorylates p53 and can enhance p53 stability. Activated p53 transcriptionally increases the expression of the p21WAF1/CIPI gene, which results in a sustained inhibition of G1 cyclin/Cdk, and prevents phosphorylation of pRb and progression from G1 into S.5 Mutations in p53 that are commonly found in solid tumors result in loss of transcriptional activity and compromised checkpoint function.
Control of the S-phase checkpoint is mediated in part by the Cdc25A phosphatase inhibiting Cdk2 activity and the loading of Cdc45 onto chromatin. If Cdc45 fails to bind to chromatin, DNA polymerase α is not recruited to replication origins and replicon initiation fails to occur.6 A more prominent mechanism for S-phase arrest is signaled through the MRN complex and the cohesin protein SMC1 by ATM.7 Loss of ATM, MRN components, or SMC1 leads to the loss of the intra-S phase checkpoint function and increased radiosensitivity. Both the CDC45 and ATM pathways represent parallel, but seemingly independent, pathways to protect replication forks from trying to replicate through DNA strand breaks. Although ATM has received the lion’s share of attention in signaling checkpoint activation in response to ionizing radiation, its family member ATR (ataxia telangiectasia and rad3-related) also plays a role in S-phase checkpoint responses.8 ATM kinase activity is inducible by radiation, whereas ATR kinase activity is constitutive and does not significantly change with irradiation. (ATR is described in more detail in Chapter 19.) In contrast to Cdc45 and ATM, ATR is probably more important in monitoring perturbations in replication that are the result of stalled replication forks to prevent the formation of DNA double-strand breaks.
The arrest of cells in the G2 phase following DNA damage is one of the most conserved evolutionary responses to ionizing radiation. It makes sense to have a final checkpoint in the G2 phase to prevent cells from entering into mitosis with damaged DNA that could be transmitted to their progeny. It follows that cells lacking the G2 checkpoint are radiosensitive because they try to divide with damaged chromosomes that cannot be aligned at metaphase to be properly apportioned to daughter cells. At the biochemical level, the regulation of the mitosis-promoting factor cyclin B/Cdk1 is the critical step in the activation of this checkpoint. At the molecular level, ATM and Chk1/2 are activated by DNA damage in the G2 phase and inhibit the activation of Cdc25A and C phosphatases, which are essential for the activation of cyclin B/Cdk1.9,10 The pololike kinase family (Plk1 and Plk3) also responds to DNA damage and can inhibit Cdc25C activation.11 A great deal of effort has been focused on the development of small molecules to inhibit checkpoint response proteins, such as Chk1, with the idea that they would inhibit radiation-induced G2 arrest and perhaps repair and thus be used as radiation sensitizers.12
DNA Repair
Ionizing radiation causes base damage, single-strand breaks, double-strand breaks, and sugar damage, as well as DNA–DNA, and DNA–protein cross-links. The critical target for ionizing radiation-induced cell inactivation and cell killing is the DNA double-strand break.13,14 In eukaryotic cells, DNA double-strand breaks can be repaired by two processes: homologous recombination repair (HRR), which requires an undamaged DNA strand as a participant in the repair, and nonhomologous end joining (NHEJ), which mediates end-to-end joining.15 In lower eukaryotes, such as yeast, HRR is the predominant pathway used for repairing DNA double-strand breaks, whereas mammalian cells use both HHR and non-HHR to repair their DNA. In mammalian cells, the choice of repair is biased by the phase of the cell cycle and by the abundance of repetitive DNA. HRR is used primarily in the late S phase/G2 phases of the cell-cycle, and NHEJ predominates in the G1-phase of the cell cycle (Fig. 13.5). NHEJ and HRR are not mutually exclusive, and both have been found to be active in the late S/G2 phase of the cell cycle, indicating that factors in addition to the cell-cycle phase are important in determining which mechanism will be used to repair DNA strand breaks.
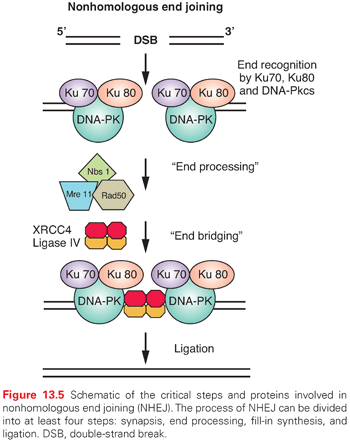
Nonhomologous End Joining. In the G1-phase of the cell cycle, the ligation of DNA double-strand breaks is primarily through NHEJ because a sister chromatid does not exist to provide a template for HRR. The damaged ends of DNA double-strand breaks must first be modified before rejoining. The process of NHEJ can be divided into at least four steps: synapsis, end processing, fill-in synthesis, and ligation (Fig. 13.6).16 Synapsis is the critical initial step where the Ku heterodimer and the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) bind to the ends of the DNA double-strand break. Ku recruits not only DNA-PKcs to the DNA ends, but also artemis, a protein that possesses endonuclease activity for 5′ and 3′ overhangs as well as hairpins.17 DNA-PKcs that is bound to the broken DNA ends phosphorylates artemis and activates its endonuclease activity for end processing. This role of artemis’ endonuclease activity in NHEJ may not necessarily be required for the ligation of blunt ends or ends with compatible termini. DNA polymerase μ is associated with the Ku/DNA/XRCC4/DNA ligase IV complex, and is probably the polymerase that is used in the fill-in reaction. The actual rejoining of DNA ends is mediated by a XRCC4/DNA ligase IV complex, which is also probably recruited by the Ku heterodimer.18,19 Although NHEJ is effective at rejoining DNA double-strand breaks, it is highly error prone. In fact, the main physiologic role of NHEJ is to generate antibodies through V(D)J rejoining, and the error-prone nature of NHEJ is essential for generating antibody diversity.
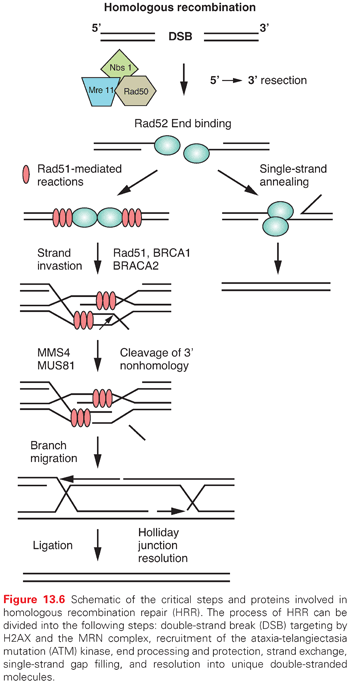
Homologous Recombination. HRR provides the mammalian genome a high-fidelity pathway of repairing DNA double-strand breaks. In contrast to NHEJ, HRR requires physical contact with an undamaged DNA template, such as a sister chromatid, for repair to occur. In response to a double-strand break, ATM as well as the complex of Mre11, Rad50, and Nbs1 proteins (MRN complex), are recruited to sites of DNA double-strand breaks (Fig. 13.6).20 The MRN complex is also involved in the recruitment of the breast cancer tumor suppressor gene, BRCA1, to the site of the break.21 In addition to recruiting BRCA1 to the site of the DNA strand break, Mre11 and as yet unidentified endonucleases resect the DNA, resulting in a 3′ single-strand DNA that serves as a binding site for Rad51. BRCA2, which is recruited to the double-strand break by BRCA1, facilitates the loading of the Rad51 protein onto replication protein A (RPA)-coated single-strand overhangs that are produced by endonuclease resection.22 The Rad51 protein is a homolog of the Escherichia coli recombinase RecA, and possesses the ability to form nucleofilaments and catalyze strand exchange with the complementary strand of the undamaged chromatid, an essential step in HRR. Five additional paralogs of Rad51 also bind to the RPA-coated single-stranded region and recruit Rad52, which binds DNA and protects against exonucleolytic degradation.23 To facilitate repair, the Rad54 protein uses its ATPase activity to unwind the double-stranded molecule. The two invading ends serve as primers for DNA synthesis, resulting in structures known as Holliday junctions. These Holliday junctions are resolved either by noncrossing over, in which case the Holliday junctions disengage and the DNA strands align followed by gap filling, or by crossing over of the Holliday junctions and gap filling. Because inactivation of most of the HRR genes discussed previously results in radiosensitivity and genomic instability, these genes provide a critical link between HRR and chromosome stability.
Chromosome Aberrations Result from Faulty DNA Double-Strand Break Repair
Unfaithful restitution of DNA strand breaks can lead to chromosome aberrations such as acentric fragments (no centromeres) or terminal deletions (uncapped chromosome ends). Radiation-induced DNA double-strand breaks also induce exchange-type aberrations that are the consequence of symmetric translocations between two DNA double-strand breaks in two different chromosomes (Fig. 13.7). Symmetrical chromosome translocations often do not lead to lethality, because genetic information is not lost in subsequent cell divisions. In contrast, when two DNA double-strand breaks in two different chromosomes recombine to form one chromosome with two centromeres and two fragments of chromosomes without centromeres or telomeres, cell death is inevitable. These types of chromosome aberrations are the consequence of asymmetrical chromosome translocations where the genetic material is recombined in what has been termed an illegitimate manner (e.g., a chromosome containing an extra centromere).
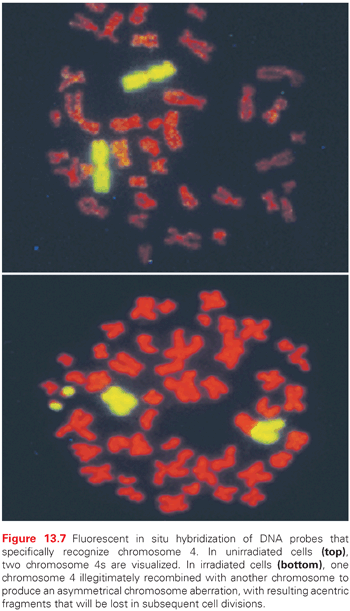
During mitosis, when a cell divides, aberrant chromosomes that have two centromeres, lack a centromere, or are in the shape of a ring have difficulty in separating, resulting in daughter cells with unequal or asymmetric distribution of the parental genetic material. The quantification of asymmetric chromosome aberrations induced by radiation is difficult and has to be performed by the first cell division because these aberrations will be lost during subsequent cell divisions. For this reason, symmetrical chromosome aberrations have been used to assess radiation-induced damage many generations after exposure because they are not lost from the population of exposed cells. In fact, symmetrical chromosome aberrations can be detected in the descendants of survivors of Hiroshima and Nagasaki, indicating that they are stable biomarkers of radiation exposure.24
Membrane Signaling
Apart from the direct of effects on DNA, radiation also affects cellular membranes. As part of the cellular stress response, radiation activates membrane receptor signaling pathways such those initiated via epidermal growth factor receptor (EGFR) and transforming growth factor β (TGF-β).25,26 Activation of these pathways promotes overall survival in response to radiation by promoting DNA damage repair and/or cellular proliferation. In addition, radiation also induces ceramide production at the membrane via activation of sphingomyelinases, which hydrolyze sphingomyelin to form ceramide. Ceramide production is linked to radiation-induced apoptosis.27
The Effect of Radiation on Cell Survival
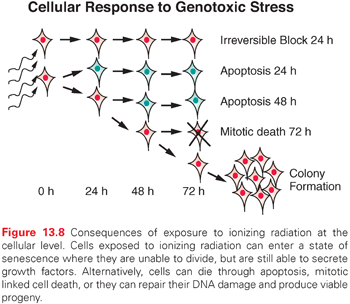
The major potential consequences of cells exposed to ionizing radiation are normal cell division, DNA damage–induced senescence (reproductively inactive but metabolically active), apoptosis, or mitotic-linked cell death (Fig. 13.8). These manifestations of DNA damage can occur within one or two cell divisions or can manifest at later times after many cell divisions.28 Effects that occur at later times have been termed delayed reproductive cell death and may also be influenced by secreted factors that are induced in response to radiation.29
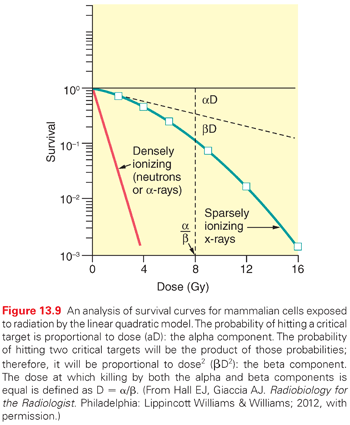
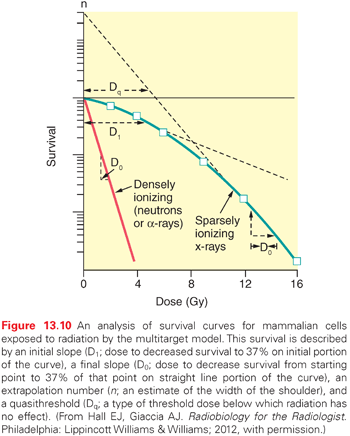
The ability to culture cells derived from both normal and tumor tissues has allowed us to gain insight into how radiosensitivity varies between tissues by analyzing the shape of survival curves. Survival curves of tumor cells often possess a shouldered region at low doses that becomes shallower as the dose increases and eventually becomes exponential. A shoulder on a survival means that these low doses of radiation are less efficient in cell killing, presumably because cells are efficient at repairing DNA strand breaks.13,14 Killing at low doses of radiation can be described in the form of a linear quadratic equation: S = e−αD−βD2 (Fig. 13.9).30 In this equation, S is the fraction of cells that survive a dose (D) of radiation, whereas α and β are constants. Cell killing by the linear and quadratic components are equal when αD = βD2 or D = α/β. Over a larger dose range, the relationship between cell killing and dose is more complex and is described by three different components: an initial slope (D1), a final slope (Do), and the width of the shoulder (n, the extrapolation number) or Dq, the quasi-threshold dose (Fig. 13.10). The extrapolation number, n, defines the place where the shoulder intersects the ordinate when the dose is extrapolated to zero, and the quasithreshold dose, Dq, defines the width of the shoulder by cutting the dose axis when there is a survival fraction of unity. In contrast to photons, the shoulder on the survival curve disappears when cells are exposed to densely ionizing radiation from particles, indicating that this form of radiation is highly effective at killing cells at both low and high doses.
In Vivo Survival Determination of Normal Tissue Response to Radiation
Although much of our knowledge on the effects of radiation on cell survival has come from cell culture studies, investigators have also devised experimental approaches to assess the clonogenic survival of normal tissues. The earliest example came from McCulloch and Till,31 who developed an assay to measure the clonogenic survival of bone marrow–derived cells in response to radiation by injecting them into a recipient mouse and quantifying the number of colonies that developed in the spleen. An analysis of these in vivo spleen assays indicated that bone marrow cells are highly radiosensitive (perhaps the most radiosensitive of all mammalian cells) in that their cell survival curve lacked a shoulder. These experiments represent two important firsts in the radiation sciences: They described the first development of an in vivo assay to assess normal tissue survival to radiation, and they demonstrated the first existence of normal tissue stem cells. Soon after, Withers and colleagues32 developed an assay to assess the survival of skin stem cells, and Withers and Elkind33 developed an assay to quantify the viability of small intestinal clonogens.
Because these ingenious approaches cannot be applied to all normal tissues, loss of tissue function instead of clonogenic survival has been used as an end point to assess radiation effects. Effects on tissue function can be grouped into the acute or late variety. Desquamation of skin by radiation is an example of an acute loss of function, whereas loss of spinal cord function is an example of a late functional effect. Acutely sensitive tissues such as skin, bone marrow, and intestinal mucosa possess a significant component of tissue cell division, whereas delayed sensitive tissues, such as spinal cord, breast, and bone, do not possess a significant amount of cell division or turnover and manifest radiation effects at later times.
In Vivo Determination of Tumor Response to Radiation
Assays have also been developed to assess the clonogenic survival of tumor cells in animals. Perhaps the most relevant of these assays is the tumor control dose 50% (TCD50) assay,34 in which the dose of radiation needed to control the growth of 50% of the tumors is determined in large cohorts of tumor-bearing animals. The TCD50 assay in animals most closely approximates the clinical situation because tumors are irradiated in animals and the ability to kill all viable tumor cells is assessed. Unlike assays in which tumor cells are irradiated ex vivo, the TCD50 assay takes into account the effects of the tumor microenvironment on tumor response. In contrast to the TCD50 assay, the tumor growth delay assay reflects the time after irradiation that a transplanted tumor reaches a fixed multiple of the pretreatment volume compared to an unirradiated control. This end point can be achieved by measuring tumor volume through the use of calipers or by a noninvasive measurement of tumor volume using bioluminescent molecules such as luciferase or fluorescent proteins. In the latter approach, all the tumor cells are stably transfected with a bioluminescent marker before implantation, and tumor growth is measured by bioluminescent activity.35 The advantage of this approach is that tumor cells can be assessed even if they are orthotopically transplanted into their tissue of origin. In another approach, tumors or cells are first irradiated in vivo, the tumor is excised and made into a single-cell suspension, and these cells are then injected into a non–tumor-bearing animal. If the cells are injected subcutaneously under the skin, the end point is tumor formation.36 If the tumor cells are injected in the tail vein of the mouse, the end point is colony formation in the lungs.37 The major advantage of these assays is that the actual number of viable cells can be determined.
FACTORS THAT AFFECT RADIATION RESPONSE
The Fundamental Principles of Radiobiology
Studies on split-dose repair (SDR) by Elkind et al.38 uncovered three of what we now recognize as the most fundamental principles of fractionated radiotherapy: repair, reassortment, and repopulation (Fig. 13.11). (Reoxygenation, described in the following paragraphs, is the fourth). SDR describes the increased survival or tumor growth delay found if a dose of radiation is split into two fractions compared to the same dose administered in one fraction. This repair is likely due to DNA double-strand break rejoining. Elkind et al. found that the survival of cells increased with an increase in time between doses for up to a maximum of about 6 hours. This finding is consistent with the clinical observation that a separation of radiation treatments by 6 hours produces similar normal tissue injury as a 24-hour separation. The shoulder of a survival curve is strongly influenced by SDR: The broader the shoulder, the more SDR and the smaller α/β ratio.
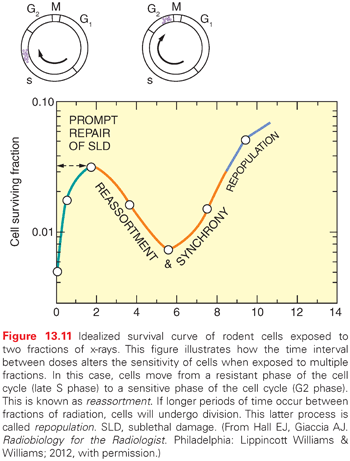
Similar to repair, reassortment and repopulation are also dependent on the interval of time between radiation fractions. If cells are given short time intervals between doses, they can progress from a resistant portion of the cell cycle (e.g., S phase) to a sensitive portion of the cell cycle (e.g., G2 phase). This transit between resistant and sensitive phases of the cell cycle is termed reassortment. If irradiated cells are provided even longer intervals of time between doses, the survival of the population of irradiated cells will increase. This increase in split-dose survival after longer periods of time is the result of cell division and has been termed repopulation. Reassortment and repopulation appear to have more protracted kinetics in normal tissues than rapidly proliferating tumor cells, and thereby enhance the tumor response to fractionated radiotherapy compared to normal tissues.
Dose-Rate Effects
For sparsely ionizing radiation, dose rate plays a critical factor in cell killing. Lowering the dose rate, and thereby increasing exposure time, reduces the effectiveness of killing by x-rays because of increased SDR. A further reduction in dose rate results in more SDR and reduces the shoulder of the survival curve. Thus, if one plots the survival for individual doses in a multifraction experiment so that there is sufficient time for SDR to occur, the resulting survival curve would have little shoulder and appear almost linear.39 In some cell types, there is a threshold to the lowering of dose rate, and in fact, one paradoxically finds an increase, instead of a decrease, in cell killing. This increase in cell killing under these conditions of protracted dose rate is due to the accumulation of cells in a radiosensitive portion of the cell cycle. In summary, the magnitude of the dose rate effect varies between cell types because of SDR, the redistribution of cells through the cell cycle, and the time for cell division to occur.
Cell Cycle
The phase of the cell cycle at the time of radiation influences the cell’s inherent sensitivity to radiation. Cells synchronized in late G1/early S and G2/M phases are most sensitive, whereas cells in G1 and mid to late S phase are more resistant to radiation.1 These differences in sensitivity during the cell cycle are exploited by the concept of reassortment during fractioned radiotherapy as well by the use of chemotherapeutic agents, which reassort cells into more sensitive phases of the cell cycle in combination with radiation.
Tumor Oxygenation
The major microenvironmental influence on tumor response to radiation is molecular oxygen.40 Decreased levels of oxygen (hypoxia) in tissue culture result in decreased killing after radiation, which can be expressed as an oxygen enhancement ratio (OER). Operationally, OER is defined as the ratio of doses to give the same killing under hypoxic and normoxic conditions. At high doses of radiation, the OER is approximately 3, whereas at low doses, it is closer to 2.41 Oxygen must be present within 10 μs of irradiation to achieve its radiosensitizing effect. Under hypoxic conditions, damage to DNA can be repaired more readily than under oxic conditions, where damage to DNA is “fixed” because of the interaction of oxygen with free radicals generated by radiation. These changes in radiation sensitivity are detectable at oxygen ranges below 30 mm Hg. Most tumor cells exhibit a survival difference halfway between fully aerobic and fully anoxic cells when exposed to a partial pressure of oxygen between 3 and 10 mm Hg.1 The presence of hypoxia has greater significance for single-dose fractions used in the treatment of certain primary tumors and metastases and is less important for fractionated radiotherapy, where reoxygenation occurs between fractions. Furthermore, most hypoxic cells are not actively undergoing cell division, thus impeding the efficacy of conventional chemotherapeutic agents that are targeted to actively dividing cells.
Although normal tissue and tumors vary in their oxygen concentrations, only tumors possess levels of oxygen low enough to influence the effectiveness of radiation killing. Although the variations in normal tissue oxygenation are in large part due to physiology governing acute changes in oxygen consumption, the variations in tumor oxygen can be directly attributed to abnormal vasculature that results in a more chronic condition. Thomlinson and Gray42 observed that variations in tumor oxygen occur because there is insufficient vasculature to provide oxygen to all tumor cells. They hypothesized that oxygen is unable to reach tumor cells beyond 10 to 12 cell diameters from the lumen of a tumor blood vessel because of metabolic consumption by respiring tumor cells. This form of hypoxia caused by metabolic consumption of oxygen has been termed chronic or diffusion-mediated hypoxia. In contrast, changes in blood flow due either to interstitial pressure changes in tumor blood vessels that lack a smooth muscle component or red blood cell fluxes can cause transient occlusion of blood vessels resulting in acute or transient hypoxia. Chronically, hypoxic cells will only become reoxygenated when their distance from the lumen of a blood vessel decreases, such as during fractionated radiotherapy when tumor cords shrink. In contrast, tumor cells that are acutely hypoxic because of changes in blood flow or interstitial pressure often cycle in an unpredictable manner between oxic and hypoxic states as blood flow changes.
Based on studies demonstrating that hypoxia can alter radiation sensitivity and decrease tumor control by radiotherapy, strategies have been developed to increase tumor oxygenation. Most importantly, it appears that tumor oxygen levels increase during a course of fractionated radiation. This may be one of the most important benefits of fractionated radiation and is termed reoxygenation (the fourth of the four Rs of radiobiology). Tumor reoxygenation during a course of fractionated radiation may also offer an explanation for the general lack of clinical efficacy of hypoxic cell sensitizers despite the clear evidence that hypoxia causes radioresistance.
Aside from using fractionated radiation, the most direct approach to increasing tumor oxygenation is to expose patients receiving radiotherapy to hyperbaric oxygen therapy. The underlying concept is that increasing the amount of oxygen in the bloodstream should result in more oxygen being available for diffusion to the hypoxic regions of tumors. Experimentally, hyperbaric oxygen therapy increases the sensitivity of transplanted tumors to radiation. The results of clinical studies with hyperbaric oxygen therapy, when combined with radiotherapy, showed improvement for two sites—head and neck cancers, and cervix cancers—but failed to show an improvement with other sites, thus calling into question its general usefulness in radiotherapy.43 In a related approach, erythropoietin (EPO), a hormone released by the kidney that increases red blood cell production, should also increase tumor oxygenation by increasing the delivery of hemoglobin-bound oxygen molecules. EPO has been effective at correcting anemia, but has not been successful in combination with radiation to control head and neck cancer and may, in fact, stimulate tumor growth.44,45
Another strategy to increase tumor oxygenation has been the combined use of nicotinamide, which increases tissue perfusion and carbogen (95% O2 and 5% CO2) breathing (accelerated radiotherapy with carbogen and nicotinamide [ARCON] therapy). Recently, a randomized phase III clinical trial demonstrated improved regional but not local tumor control in larynx cancer patients treated with nicotinamide, carbogen, and radiation versus radiation alone.46 Biologics such as antivascular endothelial growth factor (anti-VEGF) therapy have also been demonstrated to increase tumor oxygenation.47 Anti-VEGF therapy may increase tumor oxygenation by eliminating abnormal vessels that are inadequate in perfusing tumor cells—the so-called vascular normalization hypothesis. Although there is solid experimental evidence to support this hypothesis, there appears to be only a short window of time in which it could be effectively combined with radiotherapy.
Because the presence of hypoxia has both prognostic and potential therapeutic implications, a substantial effort has been invested in trying to image hypoxia.48 The goal of using imaging to “paint” radiation doses to different regions of tumors, although technically possible (as described in the next section, Radiation Physics), faces the problem that changes in oxygenation are dynamic.49 In the future, hypoxia-directed treatment may evolve from the use of hypoxic cell cytotoxins to targeted drugs that exploit cellular signaling changes induced by hypoxia such as hypoxia-inducible factor 1α (HIF-1α). However, despite the strong rationale supporting their use, at this time, there are no agents used in the clinic that target hypoxia.
Immune Response
The abscopal effects of radiation (i.e., tumor cell killing outside of the radiation field) have been attributed to the activation of antigen and cytokine release by radiation, which subsequently activates a systemic immune response against tumor cells.50,51 This response begins with the transfer of tumor cell antigens to dendritic cells and, subsequently, the activation of tumor-specific T cells and immunogenic tumor cell death. It is likely that radiation dose and fractionation influence the optimal immune response with higher doses and fewer fractions of radiation than those used in conventional fractionation schemes appearing superior in experimental models. Unfortunately, abscopal effects are uncommon because immune system evasion is an inherent characteristic of cancer cells that often dominates, even in the presence of a radiation-induced immune response. Strategies to amplify radiation-induced immune responses, and thus to overcome tumor cell evasion of the immune system, are under investigation. The combination of radiation with immune checkpoint modulators such as ipilimumab, an antibody against cytotoxic T-lymphocyte antigen 4 (CTLA-4), have shown promising, albeit anecdotal, clinical effects.
DRUGS THAT AFFECT RADIATION SENSITIVITY
For over 30 years now, chemotherapy and radiotherapy have been administered concurrently. In order to maximize the efficacy of radiochemotherapy, it is necessary to understand the biologic mechanisms underlying radiosensitization by chemotherapeutic agents. The several classes of standard chemotherapeutic agents as well as novel molecularly targeted agents that possess radiosensitizing properties will be discussed in this section.
Antimetabolites
5-fluorouracil is among the most commonly used chemotherapeutic radiation sensitizers. Given in combination with radiation, it has led to clinical improvements in a variety of cancers, including those of the head and neck, the esophagus, the stomach, the pancreas, the rectum, the anus, and the cervix. The combination of 5-fluorouracil with radiation is now a standard therapy for cancers of the stomach (adjuvant), the pancreas (unresectable), and the rectum. For other cancers such as head and neck, esophagus, or anal, 5-fluorouracil and radiation are combined with cisplatin or mitomycin C, respectively. Being an analog of uracil, 5-flourouracil is misincorporated into RNA and DNA. However, the ability of 5-fluorouracil to radiosensitize is related to its ability to inhibit thymidylate synthase, which leads to the depletion of thymidine triphosphate (dTTP) and the inhibition of DNA synthesis. This slowed, inappropriate progression through S phase in response to 5-fluorouracil is thought to be the mechanism underlying radiosensitization.52 Similar to 5-fluorouracil, the oral thymidylate synthase inhibitor, capecitabine, is also being increasingly used in combination with radiation.
Gemcitabine (2′, 2′-deoxyfluorocytdine [dFdCyd]) is another potent antimetabolite radiosensitizer. Preclinical studies have demonstrated that radiosensitization by gemcitabine involves the depletion of deoxyadenosine triphosphate (dATP) (related to the ability of gemcitabine diphosphate (dFdCDP) to inhibit ribonucleotide reductase) as well as the redistribution of cells into the early S phase of the cell cycle.53 The combination of gemcitabine with radiation in clinical trials has suggested improved clinical outcomes for patients with cancers of the lung, pancreas, and bladder. Gemcitabine-based chemoradiation has developed into a standard therapy for locally advanced pancreatic cancer. However, in some clinical trials, such as those in lung and head and neck cancers, the combination of gemcitabine with radiation has led to increased mucositis and esophagitis.54 Thus, it should be emphasized that in the presence of gemcitabine, radiation fields must be defined with great caution. Such is the case with pancreatic cancer, where the combination of full-dose gemcitabine with radiation to the gross tumor can be safely administered if clinically uninvolved lymph nodes are excluded.55 Conversely, the inclusion of the regional lymphatics in the treatment field in combination with full-dose gemcitabine produces unacceptable toxicities.56
Platinums and Temozolomide
Cisplatin is likely the most commonly used chemotherapeutic agent in combination with radiation. Although cisplatin was the prototype for several other platinum analogs, carboplatin is also frequently used in combination with radiation. Cisplatin, in combination with radiation, and sometimes in conjunction with a second chemotherapeutic agent, is indicated for cancers of the head and neck, esophagus (with 5-fluorouracil), the lung, the cervix, and the anus. Radiosensitization by cisplatin is related to its ability to cause inter- and intra-strand DNA cross-links. Removal of these cross-links during the repair process results in DNA strand breaks. Although there are multiple theories to explain the mechanism(s) of radiosensitization by cisplatin, two plausible explanations are that cisplatin inhibits the repair (both homologous and nonhomologous) of radiation-induced DNA double-strand breaks and/or increases the number of lethal radiation-induced double-strand breaks.57
Temozolomide in combination with radiation is standard therapy for glioblastoma. Temozolomide is an alkylating agent, which forms methyl adducts at the O6 position of guanine (as well as at N7 and N3-guanine) that are subsequently improperly repaired by the mismatch repair pathway. Radiosensitization by temozolomide involves the inhibition of DNA repair and/or an increase in radiation-induced DNA double-strand breaks due to radiation-induced single-strand breaks in proximity to O6 methyl adducts. Like cisplatin, temozolomide-mediated radiosensitization does not seem to require cell cycle redistribution.
Taxanes
The taxanes, paclitaxel and docetaxel, act to stabilize microtubules resulting in the accumulation of cells in G2/M, the most radiation-sensitive phase of the cell cycle. The radiosensitizing properties of the taxanes are thought to be attributable to the redistribution of cells into G2/M. Paclitaxel, in combination with radiation (and carboplatin), has demonstrated a clinical benefit in the treatment of resectable lung carcinoma.58
Molecularly Targeted Agents
Molecularly targeted agents are especially appealing in the context of radiosensitization because they are generally less toxic than standard chemotherapeutic agents and need to be given in multimodality regimens (given their often inadequate efficacy as single agents). The EGFR has been intensely pursued as a target; both antibody and small molecule EGFR inhibitors, such as cetuximab and erlotinib, respectively, have been developed. The head and neck seem to be the most promising tumor sites for the combination of EGFR inhibitors with radiation therapy. Preclinical data have demonstrated that the schedule of administration of EGFR inhibitors with radiation is important; EGFR inhibition before chemoradiation may produce antagonism.59 In a randomized phase III trial, cetuximab plus radiation produced a significant survival advantage over radiation alone in patients with locally advanced head and neck cancer.60 In a subsequent trial, however, cetuximab in combination with concurrent, cisplatin-based chemoradiation failed to produce a survival benefit in head and neck cancer patients.61 The combination of EGFR inhibitor with cisplatin-radiation requires further preclinical investigation.
Although EGFR inhibition, concurrent with radiation, is by far the best established combination of a molecularly targeted agent with radiation, other exciting molecularly targeted agents are being developed as radiation sensitizers. Targeting DNA damage response pathways is one approach to radiosensitization. Recently, agents that abrogate radiation-induced cell cycle checkpoints, such as Wee1 and Chk1 inhibitors, have been shown to radiosensitize tumor cells and are currently in clinical development in combination with chemotherapy, with clinical trials planned in combination with radiation.62,63 In addition, poly(ADP-ribose) polymerase (PARP) inhibitors have been demonstrated to preclinically induce radiosensitization, and several clinical trials combining PARP inhibitors with radiation therapy are underway.64
Other Agents
Although the most common clinically used agents in combination with radiation have been shown to produce significant clinical benefit, as described previously, other agents with different mechanisms of action have been used as radiation sensitizers as well as radiation protectors. The vinca alkaloids, such as vincristine, possess radiosensitizing properties due to their ability to block mitotic spindle assembly and, thus, arrest cells in M phase. Although vincristine is used in combination with radiation to treat medulloblastoma, rhabdomyosarcoma, and brain stem glioma, its use is principally based on its lack of myelosuppressive side effects, which are dose limiting for radiation in these types of tumors, rather than its potential radiosensitizing properties.
Also worth mention in a discussion of modulators of radiation sensitivity are agents designed to radioprotect normal tissues. One such type of drug, amifostine, is a free radical scavenger with some selectivity toward normal tissues that express more alkaline phosphatase than tumor cells, the enzyme of which converts amifostine to a free thiol metabolite. Clinical trials in head and neck as well as lung cancers have shown a reduction in radiation-related toxicities such as xerostomia, mucositis, esophagitis, and pneumonitis, respectively.65,66 However, further clinical investigations are necessary to conclusively demonstrate a lack of tumor protection and safety in combination with chemoradiotherapy regimens.
Physics of Photon Interactions
Tumors requiring radiation can be found at depths ranging from zero to 10s of centimeters below the skin. The goal of treatment is to deliver sufficient ionizing radiation to the tumor site, which can result in an absorbed dose. This involves both the availability of treatment beams and delivery techniques, and the methods to plan the treatments and ensure their safe delivery. This section will establish the general physical basis for the use of ionizing radiation in the treatment of tumors, briefly describe some of the treatment equipment, indicate physical qualities of the treatment beams themselves, and summarize the treatment planning process. Those who desire more in-depth details are referred to textbooks and other resources dedicated to medical physics and the technologic aspects of radiation oncology.67 Most patients who are treated with radiation receive high-energy, external-beam photon therapy. Here, external indicates that the treatment beam is generated and delivered from outside of the body. High-energy (6 to 20 MV) photon beams (electromagnetic radiation) penetrate tissue, enabling the treatment of deep-seated tumors. Modern equipment generates these beams with sufficient fluence to ensure delivery of therapeutic fractions of dose in short treatment sessions. Other types of particles and beams also exist for use in treating tumors both externally and internally. They are mentioned briefly later. However, as external photon beams dominate the practice (and as common basic physics principles related to delivered dose exist among the modalities), the focus here will be on photon beam generation and interactions in tissue.
As mentioned earlier, ionizing radiation kills cells via both direct and indirect mechanisms. Radiation therapy aims to instigate those ionizations and events in the tumor cells. Photons are massless, uncharged packets of energy that primarily interact with matter via electromagnetic processes. As a consequence of those interactions, an incident photon can become either entirely absorbed (giving up its energy to the ejection of an atomic electron [photoelectric effect]), or create an energetic electron-positron pair (pair production), or scatter off an electron with a reduction in energy and a change in direction and subsequent transfer of parts of its energy to the free electron (Compton scattering). The secondary electrons generated as a consequence of these interactions have residual energy, mass, and, most importantly, electric charge. They slow down in matter through multiple interactions with (primarily) the electrons of atoms, leading to excitation and ionization of those atoms. These ionizations (hence the term ionizing radiation) lead to a local absorption of energy (i.e., dose = energy absorbed per unit mass) and the direct and indirect cell killing effects necessary to treat tumors.
Thus, the use of external photon beams for cancer therapy involves a two-step process: interaction (scattering) of the photons, with subsequent dose deposition via the secondary electrons. The probability of photon interactions is energy dependent. Photoelectric interactions dominate at lower photon energies. Whereas these beams are ideal for diagnostic procedures (for their preferential absorption by tissues of differing atomic number, leading to good subject contrast), they are attenuated too quickly in tissues to supply enough interactions to be useful for therapy for any but the most superficial tumors. Pair production interactions dominate at higher photon energies; however, the probability of interacting in tissues for those high-energy photons is so low as to preclude them from general use as well. In the 10s to 100s of kiloelectron volt (keV) to the few megaelectron volt (MeV) photon energy range, Compton scattering dominates. As will be shown, these beams have sufficient penetration and can be generated with sufficient intensity to be useful for tumor treatments, especially when combined in treatment plans that comprise multiple beams entering the patient from different directions but overlapping at the tumor.
It is useful to point out physical scales of reference for external photon beam therapy. A typical megavoltage photon beam may have an average photon energy near 2 MeV. Those photons primarily undergo Compton scattering with a mean free path in tissue of approximately 20 cm. An average Compton interaction results in a secondary electron with a mean energy near 0.5 MeV (and a Compton scattered photon near 1.5 MeV, which likely escapes or scatters elsewhere in the patient). A typical secondary electron of approximately 0.5 MeV will cause excitations and ionizations of atoms as it dissipates its energy over a path length of approximately 2 mm. This could be expected to lead to approximately 10,000 ionizations, or about 5 ionizations per micron of tissue. As can be seen, therapeutic damage to the DNA of cancer cells (2 nm; see Fig. 13.2) will require very many Compton scatterings with statistical interaction among the ionizations resulting from the slowing down of the secondary electrons.
Photon Beam Generation and Treatment Delivery
As previously mentioned, effective external-beam photon treatments require higher energy beams capable of reaching deep-seated tumors with sufficient fluence to make it likely that the dose deposition will kill the tumor cells. To spare normal tissues and maximize targeting, beams are arranged to enter the patient from several directions and to intersect at the center of the tumor (treatment isocenter). Although machines containing collimated beams from high-intensity radioactive sources (primarily cobalt 60 [60
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


