INTRODUCTION
SUMMARY
Essential thrombocythemia is a clonal stem cell disorder characterized by an overproduction of platelets and associated with mutations in the JAK2, CALR, or MPL gene. Complications include thrombosis (predominantly arterial), hemorrhage, and progression to myelofibrosis or acute myeloid leukemia. Diagnosis requires exclusion of reactive thrombocytosis and other myeloid malignancies associated with a raised platelet count. Therapy is aimed at reducing thrombotic complications and includes modification of known cardiovascular risk factors and antiplatelet therapy for the majority of patients. Those at high risk of thrombosis are also considered for cytoreductive therapy with agents such as hydroxyurea, anagrelide or interferon-α. Although the majority of patients can expect to live for many years, mortality rates are increased compared to the general population as a consequence of disease complications.
DEFINITION AND HISTORY
Essential thrombocythemia (ET), one of the myeloproliferative neoplasms (MPNs), is a clonal hematopoietic stem cell disorder characterized by thrombocytosis and associated with thrombotic and hemorrhagic complications. First recognized as a specific disease entity in 19341 and as a clonal disorder in 1981,2 ET shares clinical and pathologic similarities with other MPNs, particularly polycythemia vera (PV) and primary myelofibrosis (PMF).
EPIDEMIOLOGY
The annual incidence of ET is in the order of 1 to 2.5 per 100,000 per population and appears slightly more common in females.3,4 Patients may present at any age, although ET is largely a disorder of later life with a peak incidence between the ages of 50 and 70 years. Presentation in childhood is rare but well recognized.
ETIOLOGY
Little is known about the precise etiology of this disorder, although environmental factors such as exposure to radiation have been implicated in the genesis of other MPNs.5 Both registry data and kindred studies suggest a familial tendency to develop MPNs, including ET.6,7 This predisposition appears to be explained in part by inheritance of a specific haplotype that contains the Janus family of tyrosine kinases type 2 (JAK2) gene.8
Acronyms and Abbreviations:
AML, acute myeloid leukemia; CALR, calreticulin gene; CML, chronic myeloid leukemia; ET, essential thrombocythemia; JAK2, Janus family of tyrosine kinases type 2; MPL, the thrombopoietin receptor; MPN, myeloproliferative neoplasm; PCR, polymerase chain reaction; PMF, primary myelofibrosis; PV, polycythemia vera; RARS-T, refractory anemia with ringed sideroblasts and thrombocytosis; STAT, signal transducer and activator of transcription.
PATHOGENESIS
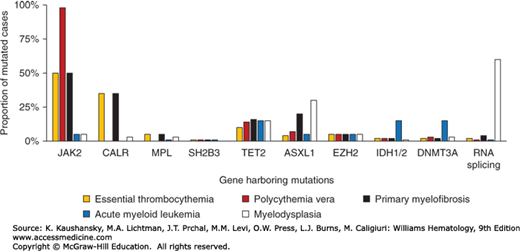
ET is characterized by hyperactive cytokine signaling which in 50 to 60 percent of cases is the result of somatic mutations targeting components of signaling pathways, including the JAK2 gene or the thrombopoietin receptor gene (MPL). Mutations in calreticulin gene (CALR) are present in the majority of JAK2/MPL–wild-type patients, leaving approximately 10 percent of ET patients without a mutation in any of these genes (JAK2/MPL/CALR–wild-type or “triple-negative” patients). A minority of ET patients also harbor mutations in transcriptional regulation pathways (Fig. 85–1).
Figure 85–1.
The spectrum and frequency of somatic mutations in myeloid neoplasms. A. Frequency of mutations in essential thrombocythemia associated with activated cytokine signaling pathways. B. Comparison of mutation frequencies in ET, related myeloproliferative neoplasms and other myeloid malignancies.
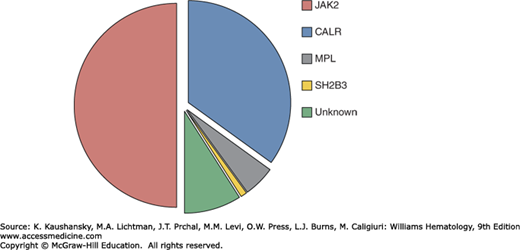
A JAK2V617F mutation is found in approximately 50 percent of patients with ET.9 JAK2, a cytoplasmic tyrosine kinase, forms a complex with and is essential for signaling by the erythropoietin and thrombopoietin receptors10,11; JAK2 also contributes to signaling by the granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, and interferon-γ receptors.12 Cytokine binding leads to a conformational change in the JAK2-receptor complex, with consequent activation of JAK2 kinase activity and recruitment of downstream signaling pathways.10,13,14 The JAK2V617F mutation alters a critical residue within the autoinhibitory pseudokinase (JH2) domain, resulting in increased JAK2 basal kinase and downstream signaling activity.15 The cellular consequences of mutant JAK2 expression include increased proliferation, cytokine hypersensitivity, cytokine-independent differentiation, and inhibition of apoptosis.9 The central role of JAK2 in erythropoiesis is highlighted by a JAK2 knockout mouse, which dies in midgestation from severe anemia.12 In addition, mice engineered to express the JAK2V617F allele recapitulate features of human ET or PV.16
Acquired mutations in MPL are found in 4 percent of ET patients, the majority of whom are JAK2–wild-type, and a similar proportion of those with PMF, but not in patients with PV.17,18 These mutations alter residues in the juxtamembrane (MPLW515) or transmembrane (MPLS505N) regions and lead to constitutive activation of the receptor complex.19,20 Expression of the MPLW515L allele in a mouse model recapitulated features of human ET and PMF.21 Occasional patients with ET, as well as PV and PMF, harbor loss of function mutations in SH2B3 which encodes LNK, an important negative regulator of JAK/STAT (signal transducer and activator of transcription) signaling.22
The majority of ET patients without a signaling pathway mutation harbor a somatic mutation in CALR (encoding calreticulin). CALR mutations are found in 15 to 35 percent of ET patients and a similar proportion of those with PMF but not in PV. Calreticulin is a key endoplasmic reticulum protein with calcium buffering and protein chaperone activity.23 Although CALR-mutant patients generally lack mutations in JAK2 or MPL, they nonetheless show signaling pathway activation, suggesting an as yet undetermined role for CALR in cytokine signaling.24,25,26
Mutations targeting pathways implicated in the control of gene transcription are found in a minority of patients with ET, as well as those with PV, PMF and other myeloid neoplasms (see Fig. 85–1B). These mutations, which may coexist with mutations in JAK2, MPL, or CALR, target genes involved in DNA methylation (TET2, IDH1/2, DNMT3A), histone modification (EZH2) or RNA splicing (SF3B1).27 In addition to its roles in cytokine signaling, JAK2 has also been shown to translocate to the nucleus where it acts as a mediator of gene transcription through the direct modification of histone proteins. Whereas nuclear translocation of wild-type JAK2 appears to be activation-dependent, JAK2 harboring a V617F substitution shows nuclear localization in the absence of cytokine stimulation.28,29
CLINICAL FEATURES
ET is often diagnosed following the incidental finding of a high platelet count, although a proportion of patients present with thrombotic or hemorrhage complications. A detailed clinical history and physical examination are necessary to exclude causes for a reactive thrombocytosis. Approximately 10 percent of ET patients have a mild degree of palpable splenomegaly at diagnosis,30 although significant splenic enlargement should raise the possibility of another MPN such as PMF or chronic myeloid leukemia (CML).
Thrombotic complications are the major source of morbidity and mortality in ET, with a prospective study indicating a cumulative incidence of 24 percent over 27 months for untreated high-risk patients.31 Arterial thrombosis predominates, affecting the central nervous system (stroke, transient ischemic attack) and cardiovascular system (myocardial infarction, unstable angina, peripheral arterial occlusion).30,31 Erythromelalgia, a distinct clinicopathologic syndrome caused by occlusion of small blood vessels, is manifest by discomfort and burning sensations in the fingers or toes, sometimes accompanied by mottling or discoloration of the skin.32 Venous events mainly comprise deep vein thrombosis and pulmonary embolism. Involvement of unusual sites such as hepatic, portal or mesenteric veins also occurs and may precede the onset of clinically overt ET (Fig. 85–2A). In one series, half of all patients presenting with hepatic vein thrombosis and a normal blood count tested positive for the JAK2V617F mutation, and a quarter of these subsequently developed a clinically overt MPN, most commonly ET.33
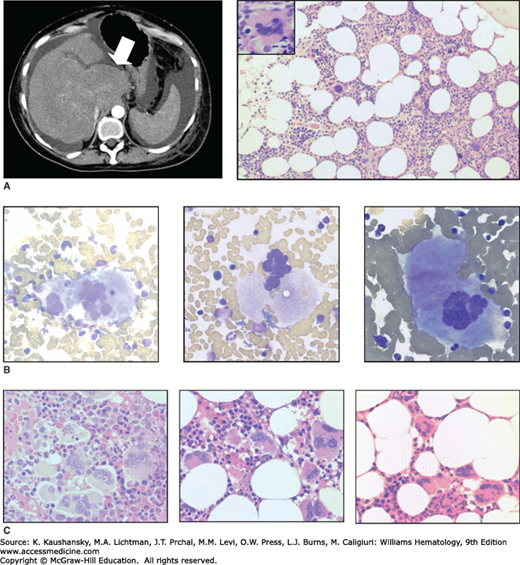
Figure 85–2.
Morphologic features of essential thrombocythemia. A. Contrast-enhanced abdominal computed tomography (CT) scan showing features of established hepatic vein thrombosis in a 53-year-old female, including hypertrophy of the caudate lobe (arrow) with atrophy of the remaining liver and surrounding ascites; the spleen is of normal size. Hematoxylin and eosin (H&E)-stained marrow trephine biopsy showing normal cellularity and increased megakaryocytes with occasional hyperlobulated forms (inset). Although the patient was JAK2V617F-positive, other investigations performed at this time, including blood count, red cell mass and cytogenetic analysis, were normal. B. Marrow aspirate from a JAK2V617F-positive essential thrombocythemia (ET) patient showing large, hyperlobulated megakaryocytes (slide stained with Wright-Giemsa). C. Marrow trephine biopsy samples from patients with ET (slide stained with H&E).
The strongest predictive factors for thrombotic complications are older than 60 years of age or a history of previous thrombosis.34 The platelet count and leucocyte count at diagnosis are poor predictors of thrombotic risk.35 Meta-analysis confirmed detection of a JAK2V617F mutation as a risk factor for both arterial and venous thrombosis.36,37 Other reported risk factors include predisposition to cardiovascular disease or increased marrow fibrosis at diagnosis.38
Serious bleeding is less common than thrombosis and mainly affects the nasal and buccal mucosa and the gastrointestinal tract, although central nervous system hemorrhage may occur.30,31 ET patients often demonstrate prolongation of the bleeding time and various abnormalities of in vitro coagulation studies, including abnormal platelet aggregation or loss of large von Willebrand factor multimers; however, the relationship of these findings to episodes of clinical bleeding is unclear.39 In a prospective study, rates of major hemorrhage were increased in patients with increased marrow fibrosis at diagnosis38 and in those with an increased platelet or leucocyte count during followup.35
Evolution to myelofibrosis is seen in a proportion of ET patients, although the reported prevalence varies widely, reflecting differences in study design, therapeutic intervention and diagnostic criteria for post-ET myelofibrosis (Chap. 86). Retrospective studies suggest that disease duration is a major predictor of progressive disease, with rates of myelofibrosis in the first decade after diagnosis of 3 to 10 percent rising to 6 to 30 percent in the second decade.34,40 The presence of marrow fibrosis at diagnosis also appears to presage progression to myelofibrosis,38 although the predictive value of other histologic features of early stage PMF, such as megakaryocyte dysplasia, remains controversial.41 Mutations in JAK2,42,43 MPL,17,18 or CALR44,45 appear to lack prognostic significance. A prospective study of high-risk ET patients indicated increased progression to myelofibrosis with anagrelide therapy, with a 5-year cumulative incidence of 7 percent for anagrelide plus aspirin versus 2 percent for hydroxyurea plus aspirin-treated patients.30 The clinical consequences of post-ET myelofibrosis are similar to de novo myelofibrosis, and the conditions are managed in the same way.
Progression to acute myeloid leukemia (AML) occurs in a small minority of patients, with retrospective studies suggesting a prevalence of 1 to 2.5 percent in the first decade after diagnosis, 5 to 8 percent in the second decade, and continuing to rise thereafter.34,40,46 Therapeutic heterogeneity in these studies, however, renders their findings difficult to interpret. Studies in PV demonstrated a significantly increased risk of AML in patients receiving genotoxic agents such as radioactive phosphorus, chlorambucil, or busulphan.47,48 The potential leukemogenicity of hydroxyurea remains controversial (see “Choice of Cytoreductive Agent” below). Importantly, transformation to leukemia has been reported in the absence of any cytoreductive therapy,49,50 indicating that AML is part of the natural history of this disorder.
Therapy of post-ET AML is often limited by the older age of the affected patients, in whom palliative treatment may be the most appropriate strategy. Overall the prognosis of secondary AML is poor (Chap. 88). Younger patients who do achieve remission with AML induction therapy may be considered for allogeneic hematopoietic stem cell transplantation.
LABORATORY FEATURES
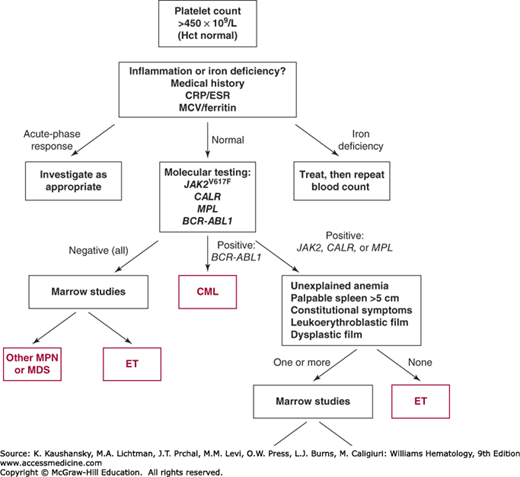
An unexplained and persistently raised platelet count generally warrants further investigation (Fig. 85–3). Establishing a diagnosis of ET requires exclusion of both reactive conditions and other myeloproliferative or myelodysplastic disorders that may present with an isolated thrombocytosis (Tables 85–1 and 85–2).
Diagnosis requires A1 to A3 or A1 + A3 to A5 A1 Sustained platelet count >450 × 109/L A2 Presence of an acquired pathogenic mutation (e.g., in JAK2, CALR, or MPL) A3 No other myeloid malignancy, especially polycythemia vera, primary myelofibrosis, chronic myeloid leukemia, or myelodysplastic syndrome A4 No reactive cause for thrombocytosis and normal iron stores A5 Marrow studies showing increased megakaryocytes displaying a spectrum of morphology with prominent large hyperlobulated forms; reticulin is generally not increased |
| CLONAL THROMBOCYTOSIS |
Essential thrombocythemia Polycythemia vera Primary myelofibrosis Chronic myeloid leukemia Refractory anemia with ringed sideroblasts and thrombocytosis 5q-minus syndrome |
| REACTIVE (SECONDARY) THROMBOCYTOSIS |
Transient thrombocytosis Acute blood loss Recovery from thrombocytopenia (rebound thrombocytosis) Acute infection or inflammation Response to exercise Response to drugs (vincristine, epinephrine, all-trans-retinoic acid) Sustained thrombocytosis Iron deficiency Splenectomy or congenital absence of spleen Malignancy Chronic infection or inflammation Hemolytic anemia |
| FAMILIAL THROMBOCYTOSIS |
| SPURIOUS THROMBOCYTOSIS |
Cryoglobulinemia Cytoplasmic fragmentation in acute leukemia Red cell fragmentation Bacteremia |
An elevated platelet count is invariably present and may be only slightly increased (e.g., ≥ 400 × 109/L) or massively elevated into the millions × 109/L. Thus, the degree of thrombocytosis varies markedly between patients. The white count may be slightly to mildly elevated but usually not above 20 × 109/L as a result of neutrophilia. The hemoglobin concentration may be normal or mildly reduced. If occult bleeding has been present, the hemoglobin may be further decreased and indications of iron deficiency may be evident in the red cells (microcytosis and hypochromia; see “Differential Diagnosis” below). Examination of the blood film often reveals large platelets which may stain poorly, and is useful in excluding features of PMF such as teardrop cells (dacryocytes) or circulating immature granulocyte precursors.
Levels of thrombopoietin are normal or slightly elevated in ET and have no diagnostic utility. ET patients may show a spurious increase in serum potassium level as a result of in vitro activation of platelets and leukocytes during processing of serum; this phenomenon can be circumvented by using a plasma sample for biochemical analysis.
Molecular testing for genetic mutations has become the investigation of choice for patients with an unexplained and persistent increase in platelet count (see Fig. 85–3). A reasonable approach is to screen all patients for the JAK2V617F mutation, following by screening for mutations in CALR and uncommon MPL in negative cases. Suitable techniques include allele-specific or real-time polymerase chain reaction (PCR) for JAK2V617F, pyrosequencing or high-resolution melt curve analysis for MPL, and fragment length analysis for CALR exon 9.25,51 In the absence of marrow cytogenetic analysis, molecular testing for the BCR-ABL1 fusion gene is also recommended to exclude CML. Screening for additional mutations (e.g., TET2) is currently of uncertain utility in routine clinical practice.
Related posts:
 Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


