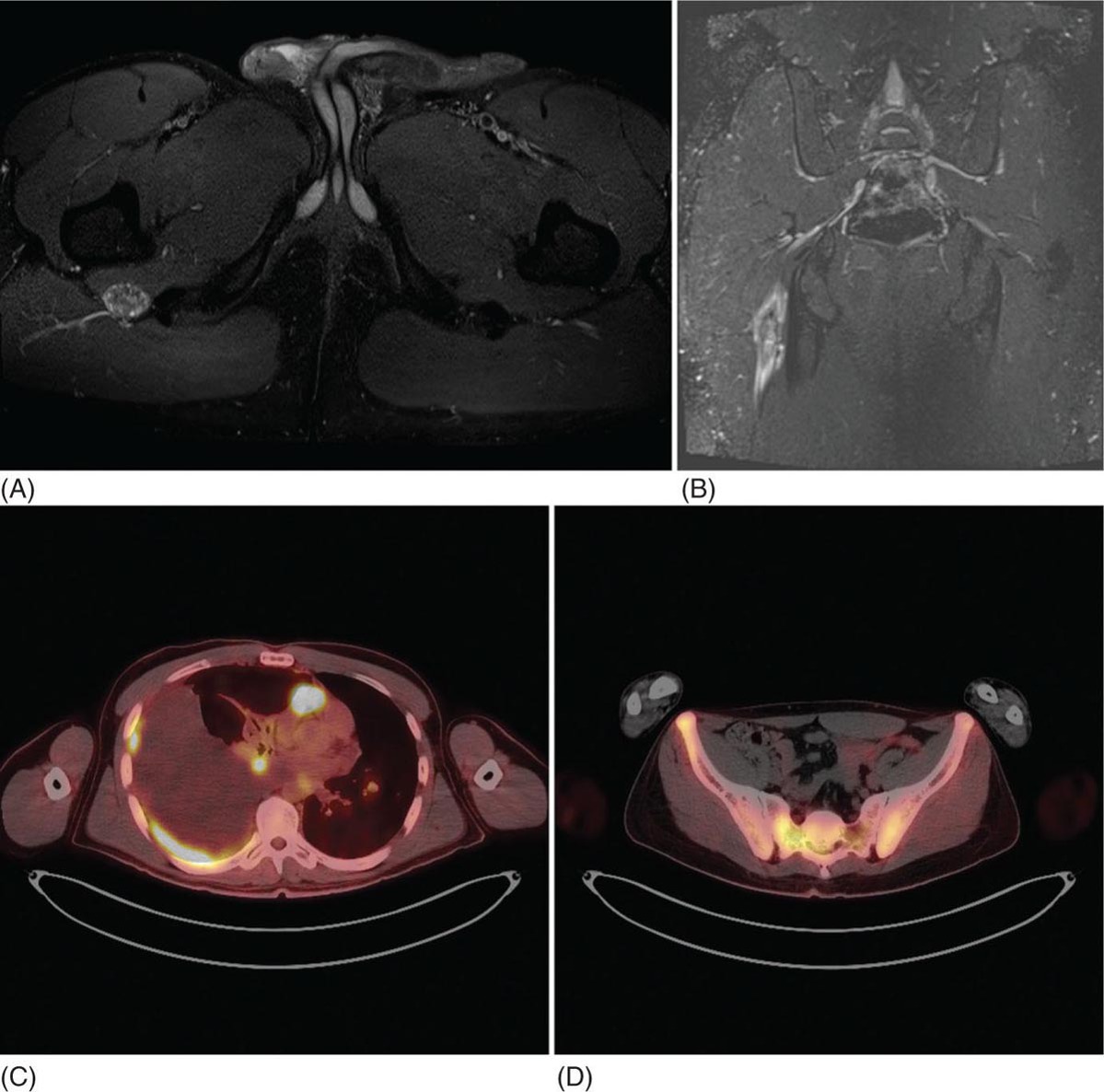
23117 Epithelioid Sarcoma Epithelioid sarcoma (ES) is an aggressive malignant soft tissue sarcoma (STS) that mainly affects young adults. It is often multifocal at presentation, with propensity for local recurrence and distant metastasis. There are two variants of this disease. One is classic ES, which usually presents as a subcutaneous or deep dermal mass in the distal extremities of young patients. The other is proximal ES, which has a predisposition for limb girdles and trunk, and a slightly later onset of presentation. Each variant has distinctive histologic features. However, both are associated with the loss of SMARCCB1/INI1 protein expression. Given the rarity of ES, the majority of our ES understanding derives from small retrospective case series and subset analysis of prospective STS trials. This chapter reviews the epidemiology, clinical presentation, diagnostic approach, molecular drivers, and treatment options for ES. Epithelioid Sarcoma, ES, SMARCB1/INI1 loss clinical presentation, diagnostic approach, epidemiology, epithelioid sarcoma, metastasis, molecular drivers, treatment options Diagnosis, Epidemiology, Neoplasm Metastasis, Sarcoma, Therapeutics INTRODUCTION Epithelioid sarcoma (ES) is an aggressive malignant soft tissue sarcoma (STS) that mainly affects young adults. It is often multifocal at presentation, with propensity for local recurrence and distant metastasis.1 It was first described as a unique entity in 1970 by Franz Enzinger in a series of 62 cases. Enzinger identified cases of soft tissue neoplasia with particular clinical and histologic behavior. These cases were previously misdiagnosed as multiple benign and malignant conditions, including chronic inflammatory process, necrotizing granulomas, synovial sarcoma, and squamous cell carcinoma.2 There are two variants of this disease. One is classic ES, which usually presents as a subcutaneous or deep dermal mass in the distal extremities of young patients. The other is proximal ES, which has a predisposition for limb girdles and trunk, and a slightly later onset of presentation. Each variant has distinctive histologic features. However, both are associated with the loss of SMARCCB1/INI1 protein expression.3 Given the rarity of ES, the majority of our ES understanding derives from small retrospective case series and subset analysis of prospective (STS) trials. This chapter reviews the epidemiology, clinical presentation, diagnostic approach, molecular drivers, and treatment options for ES. EPIDEMIOLOGY ES is a rare STS that accounts for only 0.6% to 1.0% of sarcomas, with an estimated annual incidence of 0.041 per 100,000 population.4,5 It usually presents in young adults between 20 and 40 years of age and rarely is seen in young children and older people. In children and adolescents, it is estimated to represent 5% to 8% of childhood nonrhabdomyosarcomatous STSs.6 Two variants have been described: classical type, which characteristically involves the distal extremities and affects adolescents and young adults between ages 10 and 40 (median age 33 years); and the proximal type with predilection for proximal limbs, limb girdles, and midtrunk, which presents slightly later in life, between ages 20 and 65 (median age 38 years).1,2,4,7,8 A male predisposition is noted in numerous series, with a male-to-female ratio of 1.6:1 to 1.9:1.4,8 Additionally, Caucasian race is predominant in the series. In a cohort of 441 patients from the Surveillance, Epidemiology, and End Results (SEER) database in 2008, 80% of patients were white, 12% black, and 8% reported as other. According to ethnicity, 9% were identified as Hispanic and 91% as non-Hispanic. These data are concordant with Enzinger’s original case series, in which the majority of cases were Caucasian and only a few were black.2,5,8 CLINICAL PRESENTATION AND NATURAL HISTORY ES habitually develops as indolent slow-growing lesions in the dermis, subcutaneous tissues, and deeper along the superficial fascia and tendons. Majority of tumors present as indolent firm, hard palpable masses, which are often multifocal. When located within the dermis, they present as superficial nodules that may become ulcerated 2 to 3 months after the primary lesion is noted. Ulceration ensues in about 12% of cases. These lesions are often misdiagnosed as indurated abscesses, infected ulcers, or infected sinus tracts that fail to heal despite intensive treatment. Subcutaneous lesions, on the other hand, present as small nodules described as lumps or warts that grow slowly and steady, and are not associated with limitation of function. Deep lesions occur along fascial planes, aponeurosis, and tendons, felt as multinodular or irregular masses that may move along with limb motion. Usually, deeper 232lesions are larger at presentation, and in about 25% of cases, are associated with low-grade pain. Only a minority of patients present with severe pain, motion limitation, and sensory abnormalities, which is more common in deeper lesions. Because of the absence of symptoms other than the palpated mass, patients often delay care for several months. The mean duration of symptoms prior to definitive diagnosis is around 10.5 months (range 9–30 months), and up to 30% have symptoms for more than 2 years prior to diagnosis.2,8,9 The most frequent location is the distal upper extremity (hand and forearm), which is involved in over 50% of cases. Next in frequency is the distal lower extremity, followed by proximal lower extremities, proximal upper extremity, trunk, and head and neck. Uncommon locations include the scalp, perineum, and vulva.2,5,8,9 Unlike other sarcoma, ES spreads mainly by the lymphatic system and has a tenacious propensity for local recurrence.5 Up to 70% of patients develop local recurrence, at a median length of 9.5 months after wide local excision (range 3–56 months), and over 40% of patients will have metastasis to regional lymph nodes and distant sites, most frequently to lungs. At initial diagnosis, around 47% of patients have localized disease, 28% local lymph node involvement, and 25% distant metastasis.10 Overall, patients with localized disease have substantial better prognosis than patients with regional or metastatic disease. Five-year survival rate for local disease is around 75%, for regional involvement it is 49%, and none of the patients who presented with metastatic disease were alive at 5 years. One-year survival rate for metastatic disease is around 46%. Median overall survival (OS) is about 88 months from the time of diagnosis and 5-year survival rate is about 66%.5,9,10 DIAGNOSTIC APPROACH Given the rarity of this tumor, high clinical suspicion is required for accurate diagnosis. Like any soft tissue mass, early evaluation with gadolinium-enhanced MRI is mandatory to identify features suggestive of a malignant process and the extent of tissue involvement. Referral to a multidisciplinary sarcoma center for initial management including biopsy, pathologic review, and surgical resection for localized disease is critical to improve outcomes. Initial staging should include a high-quality contrast-enhanced CT scan of the lungs, abdomen, and pelvis. PET/CT scan may be useful to identify occult lesions, particularly in advanced disease. Radiographic Features Multiple small series and case reports of radiographic characteristics are reported in the literature and support the diversity of features encountered in this tumor. On MRI, the majority of cases are of infiltrative masses. In the majority of cases, they may extend, lacking well-defined margins, and may present as multifocal lesions. In the classical subtype, some cases are seen as mainly intramuscular, while more superficial tumors have a creeping infiltrative manner extending from dermis or subcutaneous tissues deep into tendons and ligaments. Encasement of neurovascular structures and direct invasion to bone are seen in extensive tumors.11 On T1-weighted images, the majority are isointense with muscle. However, heterogeneous enhancement with areas of increased uptake are seen, corresponding to foci of hemorrhagic necrosis. Areas of central hypointensity are present in tumors with extensive necrosis. In general, necrosis is seen in up to 70% of cases and its extent correlates with tumor size, local lymph node involvement, and distant metastasis. Furthermore, necrotic nodes were seen in all patients with node involvement, but some false positives were reported. Some cases also showed areas of punctuated hyperintensity on T1-weighted images that were consistent with macrocalcifications. Similarly, T2-weighted images varied from case to case. While some tumors showed heterogeneous enhancement, others were homogeneous with different intensities compared to fat. Mild to moderate T2-weighted peritumoral edema is seen as a feathery radial pattern of nonselective high signaling on T2-weighted or STIR (short-tau inversion recovery) series.11,12 Figure 17.1 shows the radiographic findings of a patient with proximal type ES at the time of local presentation and upon progression with metastatic disease. A case report of ES showed a honeycombing pattern of fat that simulated lymphedema. Cortical necrosis was also present, exemplifying the highly heterogeneous radiologic features of this tumor.13 In the case of proximal type ES, lesions are commonly located in the chest wall, inguinal region, thigh, and perineum. By CT or MRI, these tumors were mononodular or multinodular masses with irregular indistinct borders. Calcifications were seen only in one patient of this series and were located in the periphery. It is worth mentioning that in Enzinger’s series in 1970 and subsequently in 1985, the only identified radiologic feature was the occasional presence of calcifications by x-ray exam.2,8 Low central attenuation consistent with necrosis was also common in the proximal type.14 FIGURE 17.1 Radiographic characteristics of patient with epithelioid sarcoma. (A) T2 hyperintense and enhancing lobulated tumor within the right sciatic nerve, at the level of the hamstring tendon origins. (B) STIR sequence showing significant peritumoral edema. (C) Extensive metastatic disease with multiple pleural-based lung nodules and associated large pleural effusion. (D) Diffuse osseous metastasis with iliac and sacral involvement. STIR, short-tau inversion recovery. Histopathologic Features ES is characterized by nodular aggregates of epithelioid cells immunoreactive to cytokertins (CKs), EMA, vimentin, and often CD34 positivity. There are different histologic features in the classic and proximal subtypes. In the classical form of ES, histologic features include nodular aggregates, nets, or cords of uniform epithelial cells with abundant eosinophilic cytoplasm. Nuclei have vesicular chromatin with minimal atypia and small nucleoli. Central necrosis in this subtype is prominent, giving the appearance of a granulomatous process. Epithelioid cells are located toward the center and a gradual transition to spindle morphology is seen toward the periphery. In 20% of cases, dystrophic calcification and metaplastic bone formation are seen. In the periphery, aggregates of inflammatory cells are also typical.1,4,15 On the other hand, the proximal subtype has multinodular sheets of larger, polygonal cells with higher degree of atypia with variable rhabdoid morphology. Pleomorphic epithelioid cells with large vesicular nuclei and prominent nucleoli are seen. Also, intralesional focus of necrosis is common, but the geographic pattern of the classic type with “granulomatous appearance” is absent. Rarely, a pattern with features of both classical and proximal subtypes exists. Other morphologic patterns are angiomatoid, large cell/rhabdoid, and fibroma-like. In these cases, immunohistochemistry (IHC) is key to identify the classic ES profiles.1,4 Immunophenotype by IHC is similar in classic and proximal subtypes. Both express high- and low-molecular-weight CKs with diffuse and focal patterns, most commonly, CK8, CK19, but negative or only focally positive for CK5/6. A great proportion is also positive for EMA. CD34 is positive in more than 50% of cases, which is useful to differentiate ES from carcinoma. Still, some cases show very focal or even absent CKs and CD34, and morphology may mimic other epithelial neoplasms as squamous 234cell carcinomas, benign fibrous histiocytomas, nodular fasciitis, and nonneoplastic lesions. In these cases, a high index of suspicion is important in order to assess the nuclear expression of SMARCB1/INI1 by IHC and fluorescence in situ hybridization (FISH), which is a loss in over 90% of ES.16,17 MOLECULAR AND BIOLOGIC DRIVERS There is significant genetic heterogeneity between ES tumors, with no identified pathognomonic cytogenetic abnormality. In karyotype analysis, the majority of cases have a complex karyotype with multiple numeric and structural abnormalities. ES has recurrent translocations involving the chromosome 22. The most common are t(8:22) (q22q11) and, in a minority of cases of proximal variant, t(10:22).1,18,19 These observations of recurrent involvement of chromosome 22q11 lead to the identification of the loss of two tumor suppressor genes: SMARCB1/INI1 and NF2. This loss has been documented in 20% of human cancers. However, only in rhabdoid tumors is this loss always driven by homozygous genetic inactivation of SMARCB1 gene. A report from the INT National Cancer Institute of Milan, which analyzed 40 ES cases, was the first to report the gene status in ES with loss of SMARCB1/INI1 expression. Here, homozygous gene inactivation was found for 60% of cases.7 In other SMARCB1/INI1 negative tumors, there is no consistent genetic alterations. Among STSs, complete loss of expression of SMARCB1/INI1 is found in ES, myoepithelial carcinoma, and epithelioid malignant peripheral nerve sheath tumor.1,4,20 The SMARCB1 gene encodes the adenosine triphosphate (ATP)-dependent subunit of SWI/SNF chromatin remodeling complex, which regulates genomic stability, cell cycle, and other signaling pathways. SMARCB1 is associated with activation of the Hedgehog pathway, which in embryonic life is responsible for stem cell differentiation.1,21 Furthermore, loss of SMARCB1 also results in aberrant transcription of p16INK4a and/or p21, which is a negative regulator of cyclin D1 and CDK 4/6, hence blocking phosphorylation of Rb and consequently arresting the cell cycle.22,23 Also important is the interaction of SWI/SNF chromatin remodeling complex with EZH2, a histone methyltransferase. EZH2 is highly active in stem cells, repressing genes associated with cell cycle arrest and promoting self-renewal. In adult tissues, EZH2 activity is opposed by SWI/SNF complex, which promotes differentiation and regulates self-renewal.24 The latest is important, as drugs targeting EZH2 are undergoing trials for different cancer types. It is postulated that loss of SMARCB1/INI1 is probably an earlier event causing genomic instability and explaining the complexity of karyotypes seen in ES. Other associated pathways include PI3K/AKT/mTOR and EGFR. Yet, no positive results have been yielded from targeting this pathways.1 TREATMENT CONSIDERATIONS In the localized and the metastatic setting, the rarity of ES challenges the clinical approach owing to limited evidence, which comes mainly from nonuniform retrospective series. In a series of localized ES, limitations are often due to small sample size, short follow-up periods, and inconsistency in terms of procedures and use of adjuvant treatments. From these studies, long-term survival reports range from 42% to 79%. Nonetheless, rates of local failure are considerably high: 30% to over 70% of local recurrence and over 40% progression to distant metastasis. For patients with metastatic disease, data also comes from small retrospective cohorts and small ES subset analysis from prospective clinical trials for STS. At the moment, prognosis for patients with metastatic disease is poor with 46% survival at 1 year and no patients surviving at 5 years.5 Management of the Primary Tumor Controlling localized disease consists of surgical resection with some evidence supporting the use of adjuvant RT. Only small retrospective series of patients with localized ES treated with surgery with or without RT have been published. Overall, these small retrospective series have limitations due to inconsistency regarding the surgical procedures done and the use of RT. To our knowledge, there is only one series from M.D. Anderson Cancer Center (MDACC) with consistent limb sparing surgery and use of adjuvant RT. The 10-year OS rate in this series ranged from 50% to 74%.10,25 Chase and Enzinger in 1985 published the largest series to date involving 202 patients. They reported 77% of local recurrence despite over 50% of the patients undergoing initial amputation.8 Other two small series at Massachusetts General Hospital and Memorial Sloan-Kettering Cancer Center reported that local failure rates were more than 50% and about 50% of patients subsequently required amputation as part of treatment. However, only a small proportion of patients in both of these series received 235adjuvant RT. Still, Massachusetts General Hospital series, which suggested marginal surgery with or without adjuvant RT, was associated with worse prognosis, and higher local and distant recurrence rates.10,26 Furthermore, investigators from the Mayo Clinic reported a local recurrence rate of 38% and metastatic rate of 47% after a median follow-up of 102 months. Although they found no difference in the rates of distant metastasis, they suggested that recurrence rate decreased with increasing aggressiveness of the surgery. Yet, the metastatic rate was no different between procedures.27 Older series of patients treated with only resection reported local recurrence rates of 75% to >90%.28,29 Subsequently, in 1983, Shimm et al. reported the outcomes of five patients treated with adjuvant RT. In this series, four out of the five patients had no evidence of disease at follow-up (duration of follow-up 18–84 months).30 In MDACC series of 21 patients who underwent limb sparing surgery and adjuvant RT, local recurrence was 37%, which is similar to the above-mentioned series, but all patients had limb preservation. In accordance, limb sparing surgery followed by adjuvant RT is generally recommended. Additionally, these series identified certain prognostic factors. Patients with tumors <5 cm have a 5-year overall survival (OS) rate of 79% compared with only 25% for patients with lesions >5 cm. Furthermore, OS is significantly higher, 77% for patients for primary extremity lesions compared to 39% in nonextremity locations and at 5 years, disease-free survival (DFS) rate was around 56% in patients with extremity ES versus 0% in nonextremity locations. Furthermore, necrosis more than 30%, vascular invasion, presence of local symptoms, regional lymphadenopathy, and positive margins correlate with poor prognosis.25,31,32 Probable explanations for the high rate of local recurrence despite the high proportion of aggressive surgeries is the fact that macroscopic descriptions of the procedures probably do not correlate with the microscopic surgical margins. Margin status was not reported in the mentioned series. In patients treated with local surgery and adjuvant radiation, 35% of recurrences occurred within the surgical margins and within the radiation fields. For this reason, it is recommended to attempt wide surgical margins and generous RT fields. In the MDACC series, the mean marginal field was 6.7 cm (which is the difference between the largest tumor dimension and the largest RT portal dimension). For ES a marginal field of at least 10 cm may be indicated to minimize recurrence.25 Ultimately, even though there is no well-established role for the use of sentinel node biopsy, this may be considered for prognostic implications. In cohorts of limb STS, sentinel node biopsy was positive in 17% of cases, but false negative rates of up to 29% were reported.33,34 Management of Metastatic Disease The use of systemic chemotherapy for locally advanced and metastatic ES derives from limited retrospective studies. To date, the largest cohort included 115 patients in a multi-institutional case series from 17 centers in Europe, the United States, and Japan. Cases were diagnosed between 1990 and 2016 and had available tissue for pathologic confirmation. All cases had evidence of loss of SMARCB1/INI1. Responses were evaluated by response evaluation criteria in solid tumors (RECIST). Systemic chemotherapy consisted of anthracycline-based regimens (85 patients), gemcitabine-based regimens (41 patients), or pazopanib (18 patients). In the anthracycline-based group, one patient (1%) had a complete response (CR), 18 patients (21%) partial response (PR), 45 patients (53%) stable disease (SD), and 21 patients (25%) had progression of disease (PD). Overall response rate (ORR) (CR + PR) was 22%, median progression-free survival (PFS) and OS were 6 and 16 months, respectively. In the gemcitabine-based group, two patients (5%) achieved CR, nine patients (22%) PR, 16 patients (39%) SD, and 14 patients (34%) PD. The response rate was 27%, and median PFS and OS were 4 and 19 months, respectively. Finally, in the pazopanib group, there were no CR or PR, but nine patients (50%) had SD with two prolonged SD of over 21 months; the other 50% of patients had PD. The median PFS and OS were 3 and 14 months, respectively.35 Other smaller series have also shown activity of anthracycline-based and gemcitabine-based chemotherapy as palliative treatment for ES. In a series of 21 patients, first-line palliative chemotherapy granted partial response in three patients (14%), 12 patients had SD (57%), and five patients 23% progressed. Median PFS and OS was 7 and 35 months. Both the above-mentioned series describe better responses in the classical subtype.36 Another series of 28 patients from three German participating institutions evaluating the role of palliative chemotherapy reported best response of SD in 46% of patients treated with anthracycline-based regimen. Here, PFS for anthracycline single agent was 3 months versus 8 months for combination anthracycline/ifosfamide. In the same series, the use of gemcitabine/docetaxel showed stability of disease in 83% of patients and PFS of 8 months.37 Lastly, the European Organization for Research and Treatment of Cancer (EORTC) analyzed the subgroup of metastatic ES derived from prospective EORTC trials for STS. They included ECORTC 62012 (doxorubicin vs. 236doxorubicin/ifosfamide), 62043 (pazopanib), 62072 (pazopanib vs. placebo), and 62091 (doxorubicin vs. trabectedin). A total of 27 patients were identified, 18 patients received palliative chemotherapy as first-line treatment and nine patients as second-line treatment. In the first-line setting, four patients (22%) had PR (one treated with anthracycline/ifosfamide, two treated with pazopanib, and one treated with trabectedin), 10 patients (56%) had SD (three received doxorubicin alone, six doxorubicin/ifosfamide combination, and one trabectedin), and four patients had PD. As second-line treatment, patients were treated only with pazopanib: one patient (11%) had PR, four patients (44%) SD, and four patients (44%) PD. Median PFS and OS was 4 and 11 months, respectively, for patients who received first-line treatment versus 2.7 and 9.7 months for patients who received second-line treatment.38 Most recently, tazemetostat, an oral selective inhibitor of EHZ2, was approved by the FDA for the treatment of advanced and metastatic ES. Tazemetostat efficacy was evaluated in a multicenter prospective phase II study of tumors with loss of expression of SMARCB1/INI1. As previously stated, SMARCB1/INI1 is a subunit of the SWI/SWF complex, which is a negative regulator of EZH2. EZH2 is a histone methyl transferase part of polycomb repressive complex 2 (PRC2), which functions as a key epigenetic regulator for stem cell differentiation.39 The cohort of 62 patients with ES showed an ORR of 15% (95% CI 7-26%) and disease control rate (DCR) of 26%. From the responders, over 60% had prolonged responses lasting 6 months or longer. Median OS for the cohort was 82.4 weeks (95% CI 47.4%, not estimable). Tazemetostat appears to be overall well tolerated. Most common adverse effects (AEs) are mild to moderate and include fatigue (39%), nausea (35%), and cancer-related pain (32%). Grade ≥3 AEs were reported in 10 patients (16%); most common were anemia and weight loss.40 This is the first prospective trial for patients with ES and these results are encouraging for further investigation in the adjuvant and metastatic settings. A Phase III study for its use as first line in combination with doxorubicin is currently under way (NCT04204941). SUMMARY ES is a particularly rare subtype of STS that affects mainly young adults. ES initial presentation is of slow and indolent growth with significant invasion of musculoskeletal and neurovascular tissues. Mainstay treatment approach for localized disease is surgical resection with some evidence supporting the use of adjuvant RT. Nevertheless, local recurrence considerably exceeds what is seen for other sarcoma subtypes, and distant metastasis is also common. In the scenario of locally advanced or metastatic disease, ES shows response to anthracycline, gemcitabine-based chemotherapy, and targeted therapy with pazopanib; still, responses are short lived, and the disease is ultimately fatal in all patients. In over 90% of cases, loss of expression of SMARCB1/INI1 is identified. This protein is a subunit of SWI/SWF complex, a known oncogenic driver implicated in genomic stability, regulation of cell cycle, and regulation of critical differentiation pathways. Recently, tazemetostat has shown promising activity in ES. This agent targets the inhibition of EZH2, a histone methyltransferase that is overactive in the absence of functional SWI/SWF complex. Better understanding of this molecular driver is crucial for further development of better treatment approaches for these patients.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


