Fig. 15.1
RASSF1 chromosome location (The red line on the p arm)
Buffy coat sample analysis also displayed an increased promoter hypermethylation of RASSF1A in children with thyroid carcinoma (Wong et al. 2004). Genome-wide DNA methylation profiling showed that follicular tumors of thyroid anchorage higher levels of methylation in comparison with normal thyroid tissues (Mancikova et al. 2014). Similarly, a 4.2 times hypermethylation have been observed in PTC compared to normal tissues of thyroid (Kunstman et al. 2013). These epigenetic changes were also confirmed in PTC patients when compared to adjacent thyroid tissues (ATT) (TANG and SU 2010). Comparing different forms of thyroid malignancy, this gene was detected to be hypermethylated in 71 % of thyroid carcinomas and it is more frequent in aggressive forms, which reach 80 % in undifferentiated thyroid Carcinoma (UTC) and medullary thyroid carcinoma (MTC) (Schagdarsurengin et al. 2002).
15.1.2 BRAF
V-raf sarcoma viral oncogene homolog B1 (BRAF) is a member of Raf kinase family function as an intracellular mediator of MAPK signaling pathway which is mutated in numerous cancers (Greco et al. 2009). The point mutation T1799A exchanging valine to glutamate exists in 40–70 % of PTC patients (Lee et al. 2011). This mutation causes BRAF to be consecutively and oncogenically active (Knauf and Fagin 2009). An association also found between this activating mutation and hypermethylation status and shed light on its role in the pathogenesis of thyroid cancer. It is demonstrated that tumors with BRAF mutations display a 3.6 fold incensement in methylation sites compared to normal tumors (Ellis et al. 2013). Interestingly, quantitative methylation-specific PCR (qMSP) analysis discovered an adverse correlation between BRAF mutation and RASSF1A methylation (Brait et al. 2012; Hoque et al. 2005).
15.1.3 P16
Genetic and epigenetic alterations in p16INK4a (MTS1) encodedat 9p21 region has been reported in diverse cancers. This tumor suppressor gene prevents cell progression through the G1 phase and its involvement in tumor suppression pathways makes it a significant factor in development of cancer (Bartoletti et al. 2007; Jones et al. 1996; Wang et al. 2002; Serrano et al. 2000). The two most common mechanisms of p16 inactivation are homozygous deletion or hypermethylation of the gene; it is reported that respectively 44 and 50 % of PTC and FTC patients show promoter hypermethylation in this gene (Boltze et al. 2003) (Fig. 15.2).
Fig. 15.2
P16 ribbon structure
In addition, 90 % of thyroid tumors with p16 inactivation harbor also RASSF1A inactivation (Schagdarsurengin et al. 2002). Interestingly, its expression statue changes during differentiated thyroid tumorigenesis (Ferru et al. 2006) so that it is induced in differentiated thyroid cancer and blocked during progression toward the undifferentiated status. 89 % of ATC patients lack the expression of p16 that is expressed in 46 % of malignant follicular-derived lesions of the Thyroid (Lee et al. 2008; Barroeta et al. 2006). Because of p16 over expression in follicular thyroid cells, it is believed this gene is not necessary for tumor initiation but it may be involved in progression and metastasis of thyroid cancer (Ishida et al. 2007; Ball et al. 2007; Ferenc et al. 2004; Wang et al. 2013). Although some mentioned it as a prognostic indicator (Mohammadi-asl et al. 2011; LUO et al. 2004; Gerdes et al. 2002), others observed its under or lack of expression in malignant diseases (Kiss et al. 2008; Lam et al. 2007; Huang et al. 2001) (Fig. 15.3).
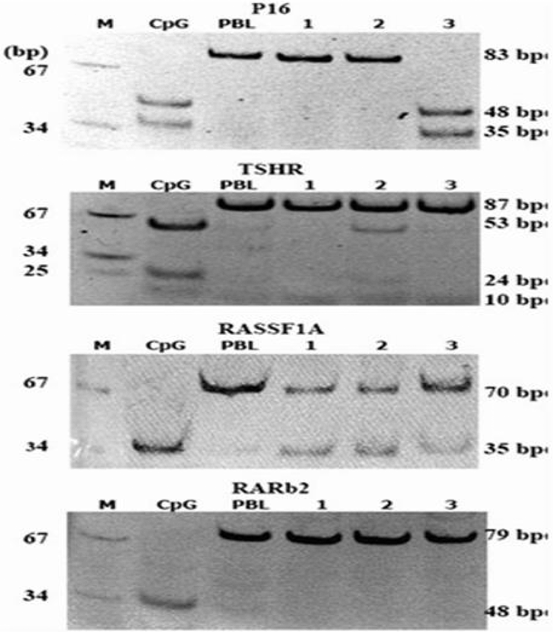
Fig. 15.3
Digestion results of PCR products of P16 (by TaqI) TSHR (by TaqI), RASSF1A (by RsaI) and RARb2 (by Bst UI) MARKER (pUC18 mSPI), CpG universal methylated DNA, PBL peripheral lymphocyte DNA. Lanes 1–3, patient’s DNA. (Mohammadi-asl et al. 2011)
15.1.4 PTEN
The tumor suppressor gene PTEN mapping on 10q 23.3 acts as a phosphatase in downstream of PI3K/Akt pathway and terminate the signaling process via dephosphorylation of PIP3. The germline mutations within this TSG cause variety of syndromes and can been seen in melanoma and thyroid cancers (Zhou et al. 2000; Xing 2008). PTEN promoter hypermethylation reported in several malignancies such as melanoma (Zhou et al. 2000), endometrial carcinoma (Salvesen et al. 2001), and thyroid cancer (Wei et al. 2013) not in gastric cancer (Sato et al. 2002). It is stated that 45.7 % of PTC patients harbor methylated PTEN promoters (Alvarez-Nuñez et al. 2006) (Fig. 15.4).
Fig. 15.4
PTEN ribbon structure
15.1.5 RARβ
Retinoic acid receptors consist of two family as retinoid X receptors (RXRs) and retinoic acid receptors (RARs), which are necessary for transduction of retinoid signals. Each of these nuclear hormone receptor families contains α, β and γ isoforms involving in suppression of PI3K/Akt pathway through either PI3K or phosphatase PTEN targeting (Bastien and Rochette-Egly 2004). Tumor suppressor gene retinoic acid receptor-beta2 (RARβ2) encoded at 3p24 (Wu et al. 2006) exists in a hypermethylated condition in several cancers including 22 % of PTC patients (Cras et al. 2007; Li et al. 2014; Russo et al. 2011). Since no mutation reported in its relation to cancer, epigenetic control by methylation, and loss of heterozygocity have been considered as inactivation processes. Evaluation of type I iodothyronine 5’-deiodinase (5’ DI) documented that respond to retinoid therapy using retinoid acid (RA) may correlate with disregulation of expression in RA receptors including RARβ (Schmutzler et al. 2004). In addition, there is a close relationship between methylation of tazarotene-induced gene 1 (TIG1), also known as RAR-responsive 1 gene, and RARβ methylation confirming that induction of TIG1 occurs in a RAR-specific manner (Zhang et al. 2004). Interestingly, retinoic acid-resistant thyroid cancer cells called FTC238 harboring the same methylation pattern in RARβ, compared with retinoic acid-sensitive thyroid cancer cells (FTC133), which bear hypermethylated histones on RARβ promoter (Cras et al. 2007). Furthermore, the expression of RARs suggested to be considered as an indicator for distinguishing adenoma and well-differentiated thyroid carcinoma (Gauchotte et al. 2013). Methylation of RARβ has been shown to be chemically modified; epigenetic modifiers such as nickel subsulfide (Ni3S2) (ZHANG et al. 2011) and PD153035 (Grunt et al. 2005) cause hyper- and hypomethylation of RARβ, respectively. Interestingly, re-expression of RARβ2 using demethylating agent 5-aza-2-deoxycytidine causes a significant inhibition of thyroid cancer cell growth (Miasaki et al. 2008). A study showed that smoking, which is a well-known risk factor in malignancies, induces an aberrant hypermethylation in RARβ and cause thyroid tumorigenesis (Kiseljak-Vassiliades and Xing 2011) (Fig. 15.5).
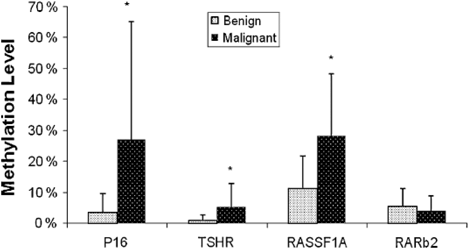
Fig. 15.5
Quantitative methylation level of four genes in benign and malignant thyroid tumors. (Mohammadi-asl et al. 2011)
15.1.6 The 14-3-3σ
14-3-3σ also named human mammary epithelium-specific marker 1 (HME1) , is a cell cycle regulator which acts as a G2/M arresting molecule expressing in malignant thyroid tissues (Lal et al. 2008). Interestingly, 14-3-3σ observed to be expressed in all papillary carcinomas but not follicular carcinomas and adenomas, and it proposed that this protein might not be required for development of thyroid follicular tumors (Ito et al. 2003). In addition, it has been postulated that this molecule that expresses under direct activation of p53, is induced after DNA damaging and plays a significant roles during several kind of malignancies including breast, prostate, skin and colon cancers (LODYGIN and HERMEKING 2005). Epigenetic analysis suggested that 14-3-3σ expression in thyroid cancer cell lines is epigenetically controlled by an alteration in CpG methylation (Lal et al. 2008). More studies are required to elucidate its function in thyroid malignancies.
15.1.7 TSHR
Thyroid-stimulating Hormone Receptor (TSHR) also known as LGR3, CHNG1 or hTSHR-I located on 14q31 is a membrane protein and a major regulator of thyroid cell metabolism. TSHR protein is a receptor for thyrothropin and thyrostimulin, which its activity is mediated by adenylate cyclase. (http://www.ncbi.nlm.nih.gov/gene/7253). Methylation of CpG islands in 5” flanking areas of the gene is known to be associated with carcinogenesis, development, progression and decreased tumor recurrence in PTC (Xu et al. 2010; Dai et al. 2010; Smith et al. 2007) and introduced as a diagnostic marker of malignancy to distinguish FTC from benign adenoma (Xing et al. 2003b). In addition, this methylation pattern occurred more preferentially in undifferentiated thyroid carcinoma (UTC) compared with PTC and FTC (Schagdarsurengin et al. 2006). Like previous genes, there is an association between T1799A BRAF mutation and TSHR methylation in PTC (SHI et al. 2009). It is also shown that transcription of TSHR is regulated through a methylation-dependent mechanism in Rat (Yokomori et al. 1998) (Fig. 15.6).
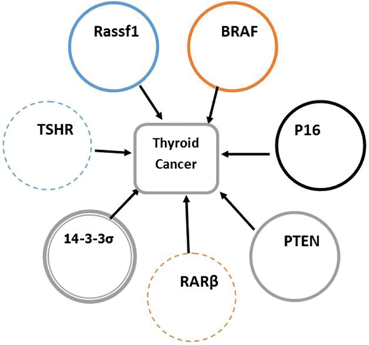
Fig. 15.6
Thyroid cancer and candidate genes
15.1.8 Other Genes
Growing epigenetic studies have updated the list of genes, which are epigenetically controlled during thyroid carcinogenesis (Table 15.1). Further researches are needed to clarify the role of epigenetic regulation in thyroid malignancy (Table 15.2).
Table 15.1
Comparison of the methylation status in candidate genes
Methylated genes in PTC patients (%) | Reference | |||
|---|---|---|---|---|
P16 | TSHR | RASSF1A | RARβ2 | |
60 | 72 | 24 | 48 | (Mohammadi-asl et al. 2011) |
44 | – | – | – | (Boltze et al. 2003) |
41 | – | – | – | (Lam et al. 2007) |
27 | – | – | – | (Wang et al. 2013) |
25 | – | 62 | – | (Schagdarsurengin et al. 2002) |
– | 41.1 | – | – | (Xu et al. 2010) |
– | 59 | – | – | (Smith et al. 2007) |
– | 68 | – | – | (Dai et al. 2010) |
– | 43 | – | – | (XiaoGuang et al. 2009) |
– | – | 20 | – | (Xing et al. 2004) |
– | – | 32 | – | (Nakamura et al. 2005) |
– | – | 62 | – | (Schagdarsurengin et al. 2010) |
Table 15.2
Other genes in thyroid cancer
Gene | Position | Methylation Site | Type of Thyroid Cancer | Expression status | Reference |
|---|---|---|---|---|---|
MT1G CRABP1 | 16q13 15q24 | 5-UTR | PTC | Down-regulated | (Huang et al. 2003) |
HBB | 11p15.5 | 5-UTR | ATC | Down-regulated | (Onda et al. 2005) |
HNF3β/FoxA2 | 20p11 | 5-UTR | ATC PTC | Down-regulated | (Akagi et al. 2008) |
PKG | 17q21 | 5-UTR | PTC | Down-regulated | (Han and Kim 2009) |
TSH-α | 6q12 | 5-UTR | PTC | Up-regulated | (Han et al. 2009) |
SLC26A4 | 7q31 | 5-UTR | various | Down-regulated | (Xing et al. 2003a) |
FHIT | 3p14.2 | 5-UTR | DTC | Down-regulated | (Yin et al. 2010) |
TTF-1 | 9q34.13 | 5-UTR | UDC(ATC) | Down-regulated | (Kondo et al. 2009) |
SERPINA5 | 14q32.1 | 5-UTR | PTC | Down-regulated | (Lee et al. 2013) |
RASSF2 | 3p21.3 | 5-UTR | Various cell lines | Down-regulated | (Schagdarsurengin et al. 2010) |
RIZ1(PRDM2)
Related posts: Reduced Risk of Cancer in Schizophrenia, a Bridge Toward Etio-Pathology and Therapy of Both Diseases Reduced Risk of Cancer in Schizophrenia, a Bridge Toward Etio-Pathology and Therapy of Both Diseases
 Biodynamic Phenotypic and Epigenetics Changes of Circulating Tumor Cells: Their Application in Cancer Prognosis and Treatment Biodynamic Phenotypic and Epigenetics Changes of Circulating Tumor Cells: Their Application in Cancer Prognosis and Treatment
 An Introduction to Impact of Bio-Resonance Technology in Genetics and Epigenetics An Introduction to Impact of Bio-Resonance Technology in Genetics and Epigenetics
 Malignant Rhabdoid Tumor: Epigenetic Mechanism of Tumorogenesis Malignant Rhabdoid Tumor: Epigenetic Mechanism of Tumorogenesis
 Sentinel Gene Within Cell Territory and Molecular Platforms in Cancer: Methylation Diversity of p53 Gene in Brain Tumors Sentinel Gene Within Cell Territory and Molecular Platforms in Cancer: Methylation Diversity of p53 Gene in Brain Tumors
 Exploring ATM and Methylation in Cancer: Emphasizing on Brain Tumors Exploring ATM and Methylation in Cancer: Emphasizing on Brain Tumors
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|