Fig. 11.1
Histone acetylation. HA Histone acetylation, HAT Histone acetyltransferases, HDAC Histone deacetylases
11.1.4 Histone Methylation
Histone Methylation (HM) also occurs at lysine-(K) and arginine-(R) residues without changing the chromatic architecture, but is active as binding sites for the alternative proteins that may be involved in chromatin condensation (Nielsen et al. 2001). Editing of methyl marks (MM)is performed by S-adenosylmethionine (SAM)- dependent methyltransferases(13- Tsukada et al. 2006) and erasing of MM by either the Jumonji family of demethylases or lysine-specific histone demethylases 1 (LSD1) and 2 (LSD2) (Shi et al. 2004). Arginine methylation of histone proteins is known to antagonize the alternative histone marks, followed by raising the histone code complexity (Guccione et al. 2007). A brief insight of HM-process is provided (Fig. 11.2).
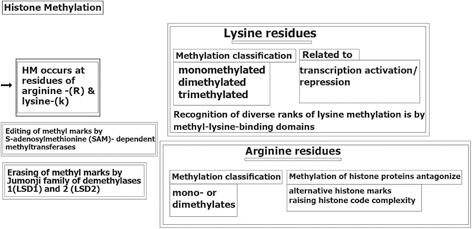
Fig. 11.2
Histone methylation. HM histone methylation, (LSD1) and 2 (LSD2) lysine-specific histone demethylases 1
Epigenetic modification play a key role in cancer development. In this platform, genomic hypomethylation is considered as a global event, and CPG methylation is responsible for silencing of tumor suppressor genes (Esteller 2008; Dobrovic and Simpfendorfer 1997; Deng et al. 1999; Hatzimichael et al. 2009; Hatzimichael et al. 2012; Esteller 2000, 2006) (Fig. 11.3). In addition, there are complementary events such as distraction of the histone modification territory, and histone modifiers include mutation of deacetylases, and amplification of methyltransferases and demethylases (Fraga et al. 2005; Ropero et al. 2006; Cloos et al. 2006). At a glance there are three major and interactive platforms in cancer and epigenetic modifications; (1) genomic hypomethylation, (2) CPG island, as a contrast territory against platform 1, and (3) Distraction of Histone modification territory (Fig. 11.3).
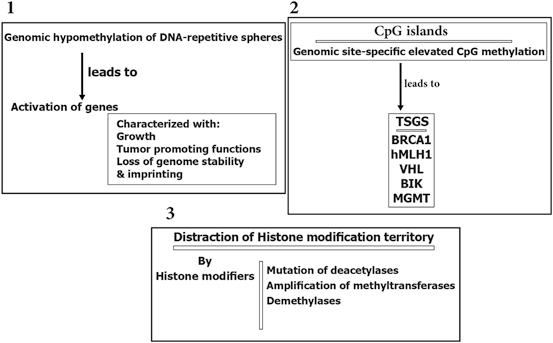
Fig. 11.3
Epigenetic modification in cancer
Furthermore, epigenetic reservoir may be altered through protein activation or inactivation.
More specifically, the epigenomic stabilization and instabilization relies on enzymatic territory and could be predisposed to mutation in different malignancies (Hatzimichael et al. 2013; Yan et al. 2011; Simo-Riudalbas et al. 2011; Ley et al. 2010; Thol et al. 2011; Delhommeau et al. 2009; Weissmann et al. 2012) (Fig. 11.4). Besides, the metabolic genomic mutations are reported for isocitrate dehydrogenase (IDH) 1 and IDH2 genes in patients with myelodysplastic syndromes (MDS) and acute myelocytic leukaemia (AML) (Ward et al. 2010). In another study in AML, IDH1 and IDH2 mutations revealed to have common epigenetic defects with TET2 mutations. However, specific hypermethylation signature has been found in AML patients with mutations of IDH1/2 (Figueroa et al. 2010). Interestingly, influential mutation on the Polycomb repressive complex (PRC), per se EZH2, are capable to modify histone elements. Moreover, EZH2 overexpression are found in the epithelial neoplasms and leukemia (Varambally et al. 2012; Benetatos et al. 2013; Simon and Lange 2008). Diversiy in the type of mutations include activating mutations of EZH2 in B-cell lymphomas, missense, nonsense, and frameshift mutations in myeloid malignancies (Sneeringe et al. 2010; Ernst et al. 2010; Nikolosk et al. 2010). Furthermore, silencing of tumor suppressor genes (TSGs) by promoter CpG methylation in breast tumorigenesis has been reported. They have highlighted that the TSG methylation in BCcould be used as a potential marker for cancer management. In a review article the epigenetic alterations in BC, biological and clinical insinuation are provided (Xian et al. 2013).
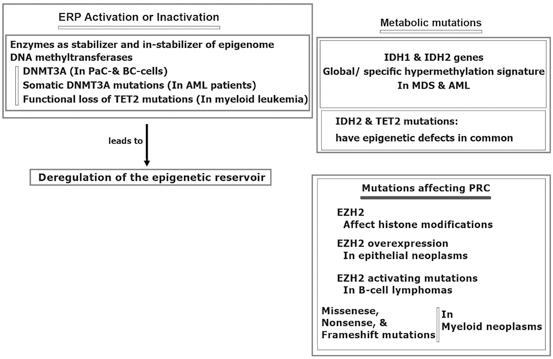
Fig. 11.4
Epigenomic regulation in cancer. AML acute myelocytic leukemia MDS myelodysplastic syndrome BC breast cancer, DNMT3A DNA methyltransferase gene, ERP epigenetic regulatory proteins, EZH2 histone-lysine N-methyltransferase, IDH isocitrate dehydrogenase
By considering the multistep carcinogenesis, divese capability of tumor cells through initiation and progression does not, solely, rely on the genetic alterations. But, epigenetic changes assist and enable the neoplastic cells to behave as the cancer cells in malignant phase with the possible reversible capacity (Fig. 11.5). However, still there are some unmasked facts in cancer epigenetics which require the harmonic and complementary approaches by considering the clinical follow-up data.
Fig. 11.5
Cancer epigenetic. MDS myelodysplastic syndromes, PaC pancreatic and, PRC polycomb repressive complex, TSGS transcriptional tumor suppressor genes silencing, TET2 tet methylcytosine dioxygenase 2
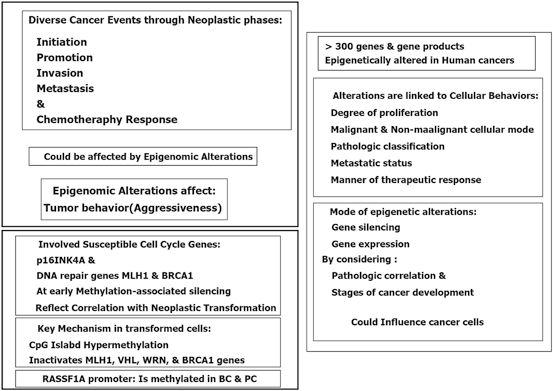
11.2 Retinoids
Retinoids with capability of antiproliferative and proapoptotic impacts, are involved in the decisive biological processes and also act as the suppresser of carcinogenic development. Retinoic acid (RA), as a derivative of vitamin A or retinol, has a remarkable epigenetic regulatory role in gene transcription (Chambon 1996). It has also a crucial impact on the morphogenic progress and development of postpuberty mammary gland (Montesano et al. 2002; Wang et al. 2005). Retinoids and their products induce differentiation in various cell types as well.
Retinoids and all-trans retinoic acid, 13-cis retinoic acid, bexarotene as the Retinoids’ natural metabolized and synthetic products have the key role in cell differentiation. Retinoids apply their actions by binding to the nuclear retinoic acid receptors (α, β, γ), in those the regulatory impact is due to transcriptional and homeostatic characteristics and their role in neoplastic transformation. By considering the retinoids’ efficacy in cancer prevention and treatment, an achievement is the treatment of of leukemic subtypes characterized with chromosomal translocations. So far, the therapeutic limitation in the prevention and treatment of solid tumors may be due to the epigenetic silencing of retinoic acid receptor beta (RARβ) which could be developed by including assessment of RARβ and downstream genes in the solid tumor (Bistulfi et al. 2006; Connolly et al. 2013). Moreover, the “dualistic role” of the retinoic acid signaling pathway in cancer is reviewed (Coyle et al. 2013). They have emphasized on the quadro-radial factors including gene transcription, interactions with other transcription targets, apoptotic pathways, and the immune machinery. It was also referred to the therapeutic impact of retinoid on innovation of an appropriate cancer therapy. Then it could be stated that retinoic acid could be considered as a hero in cancer.
Retinoids are the promising targets in cancer prevention and treatment, especially in breast cancer. Partly, the therapeutic success in the prevention and treatment of solid tumors may may be due to the epigenetic silencing of RARβ (Connolly et al. 2013).
Retinoic acid receptors (RARs) belong to the nuclear hormone receptor superfamily, and are characterized with the heterodimeric partners, known as the retinoid X receptors (RXRs).
They are capable to regulate genes having a direct repeat five (DR5) retinoic acid response element (RARE) within the promoter regions. Regarding the pharmacologic levels of retinoid-derived ligands, all-trans retinoic acid (AT-RA) and 9-cis-retinoic acid, for RARs and RXRs are respectively aimed to transactive the heterodimeric partners. In addition, the reduction of RARβ2 or RARβ4 mRNA is reported in breast cancer cell lines (Hoffmann et al. 1990; Swisshelm et al. 1994). Besides, Reduction or lack of the RARβ2 mRNA expression in primary breast cancers have been also reported (Widschwendter et al. 1995; Zhang et al. 1994).
The biological effects of RAR are summarized (Evans and Kaye 1999; Chambon 1996; Altucci and Gronemeyer 2001; Balmer and Blomhoff 2002):
1.
Retinoids, including retinoic acid (RA) regulate expression of genes and involved in cell proliferation, differentiation, and apoptosis. They play important role in normal development of embryon and health status of adult.
2.
The multiple influential behaviors of retinoids are mediated by RARs and RXRs.
3.
To guarantee the biological impact, the liganded RAR binding to RARE will lead to the expression or suppression of Retinoid target gene.
4.
RA generates antiproliferative effects in tumor cells.
5.
RARs directly mediate RA effects by regulating of gene expression.
Epigenetic silencing could repress the transcriptional function which lead to the reduction of RARβ2 expression (Xu et al. 1994; Houle et al. 1993; Liu et al. 1996). It is also reported that the RARβ-prime as a truncated, oncogenic RARβ protein is entirely expressed in breast cancer cell lines. Its worth to highlight that this isoform could block the functions of tumor suppressor of RARβ2 and RARβ4 protein isoforms as well (Lee et al. 1995; Sabichi et al. 1998). RARβ2 as a two edged sward, is capable to activate either tumor suppressor or antimetastatic programming. However, “the RARβ2 could be defined as a reflective mirror with multi-potential territory which facilitates diverted interactions with variety of targets at biological and molecular levels” (Mehdipour et al. 2012).
Interstingly, an incorporated process is essential through the RA receptor α (RARα), RA signal elicit chromatin modifications in RA signal leads to the transcription of the RA receptor β2 which is located at chromosome 3p24 (Chambon 1996; Dilworth and Chambon 2001). Furthermore, RARβ2 maintain an independent transcription accompanied by only few downstream RA-responsive genes (Brand et al. 1988; Sucov et al. 1990; Husmann et al. 1991). Retinoids have also multi-influential capacities, however its informative characteristics is summarized (Fig. 11.7) (Mehdipour et al. 2012).
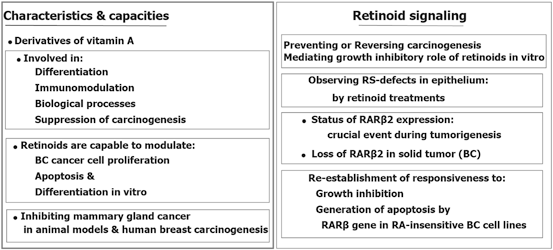
Fig. 11.7
Multi-influential capacities of retinoids
In RA-sensitive BC cell line, an inactive mode of transcription was found for RARβ2 in which notable value of repressive chromatin modifications including DNA hypermethylation could be characterized. Besides, simultaneously, with the conversion of RARβ2 alleles from a permissive transcriptional status into a nonpermissive mode which is characterized by aberrant DNA hypermethylation, cells are converted to RA resistance (Ren et al. 2005). In addition, numerous proteins involved in retinol or RA metabolism found as unbalanced or down-regulated in breast cancer cells (Andreola et al. 2000; Guo et al. 2000; Mira-y-Lopez et al. 2000; Rexer et al. 2001), including retinol binding protein 1 (CRBP1) which is involved in the morphological differentiation of human breast epithelial cells (Farias et al. 2005). The CRBP1 is encoded by the RBP1 gene, was characterized as a downstream RAR-regulated gene, as one of the RAR targets (Smith et al. 1991; Husmann et al. 1992). RARβ2 is also involved in transcription of RA-responsive gene and retinoid signaling (Pozzi et al. 2006; Kawakami et al. 2005; Vermot et al. 2005a, b). RARβ has four isoforms with different biological functions of those RARβ2 play an influential capacities in carcinogenesis and therapy and could be listed as cancer preventive and therapeutic agents in BC. The characteristics of RARβ2 is summarized in Fig. 11.8.

Fig. 11.8
The characteristics of RARβ2
Ectopic RARβ2 expression regulate downstream factors which are presentive of differential expression of transcription factors, signalling molecules and metabolic enzymes (Feinberg et al. 2006). RARβ2 is also capable to change the gene expression pattern of AT-RA in cell lines (Kurbel 2013). In addition, the ectopic RARβ2 expression, the type and origin of cell may have role in characterizing the expression profile. Besides, an interactive and regulatory manner may be involved between mode of RARβ2 expression and multi- pathways. So a global genetic programming may be involved for managing the status of gene regulation. In this regard, three upregulated genes by RARβ2 on chromosome Xq28 have been reported at cell line level including ARHGAP4 as a GTPase-activating prote, RPL10 as a 60S ribosomal proteand SSR4 as signal sequence receptor delta 4 (Tribioli et al. 1996; Oh et al. 2002; Mangelsdorf et al. 1994; OMIM: 300090). However, the importance of X chromosome may be due to the involvement of interactive targets in this chromosome with beta retinoic acid. Furthermore, the highlighted points of a review on RARβ2 gene in breast cancer is presented in Tables 11.1, 11.2, 11.3 and 11.4 (Mehdipour et al. 2012).
Table 11.1
Distinctiveness and mechanisms of of RAR-β isoforms. (Adapted from Mehdipour et al. 2012)
Name of elements | Characteristic of element/mechanisms | Associated with/via/related to/influenced by | Advantages/disadvantages |
|---|---|---|---|
RXR | Reduce the mucocutaneous toxicity | Associated with retinoid treatment | – |
4-HPR | Inhibits proliferation of BC- cells without capacity to express RARs | Via retinoid receptor-independent mechanisms | Favorable preclinical outcome |
Induce its inhibitory effects | Related to its reduced toxicity compared with other retinoids | Limited therapeutic value in patients with advanced BC | |
Induce RAR transcriptional activation and repression in breast cancer cells | – | – | |
Preferentially accumulates in breast tissue | – | – | |
ATRA RARS | Inhibits the proliferation of BC cells is by inducing G1 cell cycle arrest | Sensitivity of ER-positive BC cells to the growth inhibitory effects of ATRA (by expressing RAR a), but ER-negative cells are commonly resistant (due to lacking or small amount of RARa) | |
RAR- β antagonist & expression of RAR-β antisense could inhibit ATRA sensitivity of ER-positive cells |
Table 11.2
The characteristics of some environmental/dietry factors in epigenetics. (Adapted from Mehdipour et al. 2012)
Factors | Characteristic of element | Involved mechanism | May lead to | Influenced by |
|---|---|---|---|---|
Dietary methionine | Vital amino acid (dietary methionine) | Cytosine methylation/CpG methylation patterns | Normal genome-wide DNA methylation patterns | Dietary agents |
Transposons | Transposable elements/mobile genetic | Heavily methylated/transcriptionally silent in somatic cells/disturbed DNA methylation | Genetic mutations/transcriptional defect/altered establishment and maintenance of epigenetic status | Cellular stress/environmental and dietary agents |
Dietary chemopreventive agents: butyrate, diallyl disulfide, and sulforaphane | – | HDAC inhibitory activity | – | Dietary agents |
Resveratrol | A member of sirtuin family of NAD-dependent deacetylases | An inhibitor of SIRT1, a member of the sirtuin family of NAD-dependent deacetylases | Improves health and extends life span | Dietary agents |
Green tea polyphenols and phenethyl isothiocyanate | Dual actions of DNMT and HDAC as inhibitors in cancer cells/on DNA and chromatin | Epigenetic modifiers | Cancer prevention | Dietary intervention |
Table 11.3
The functions of chemopreventive agents in epigenetic. (Adapted from Mehdipour et al. 2012)
Name of chemopreventive agent | Alternative name/derivates | Function | Disadvantages | Advantages |
|---|---|---|---|---|
Natural retinoids: retinoid 9-cis retinoic acid (9-cis RA, alitretinoin) | ATRA | Inhibit growth of BC- cells | Lacking clinical efficacy | |
Transactivates | Toxic side effects: Hyperlipidemia, mucocutaneous, liver toxicity | |||
RARs and RXRs | ||||
Binds RAR | ||||
Does not bind RXR | ||||
Synthetic N.R.-derivatives: The 9-cis RA, 4-HPR (fenretinide) | Synthetic derivative of ATRA | Inhibit growth of BC- cells | Higher potency | |
Transactivate RAR responsive genes | Less toxicity | |||
Have significant RARs binding | ||||
Synthetic retinoids: LGD1069 (bexarotene, Targretin) | Synthetic derivative of 9-cis RA, 4-HPR | Inhibit growth of BC- cells | No significant binding RAR | Have selective binding of RXRs |
Transactivates | No transactivation of RAR responsive genes | Higher potency | ||
RXRs | Less toxicity | |||
ATRA & Cell Cycle | Interaction between cell cycle regulators with antiproliferative effects of ATRA in BC cells | (1) decreased expression of cyclin D1 and D3, (2) activity of cdk2 and cdk4, and (3) expression and phosphorylation of pRb could be associated with growth inhibition induced by ATRA in BC cells | ||
Retinoids/RAR- β gene | 1-Retinoids alter gene expression in target cells | Important event in tumorigenesis may be loss of RAR- β 2 mRNA expression | ||
2- RAR β gene may be considered as a tumor suppressor | ||||
RAR-β 2 | 1-Reduced RAR-β 2 mRNA expression has been observed in BC | 1-RAR-β expression mediates the growth inhibitory effects of retinoids | RAR- β b was induced in 33 % of BC patientstreated with ATRA for 3 weeks | |
2-RAR-β transcription is downregulated in BC cell lines and tumors, but upregulated in normal mammary epithelial cells | 2- RAR-β2 mRNA induction is associated with growth inhibition in response to ATRA | |||
3-resistance to ATRA could be associated with a collapse in RAR- β2 inducibility in BC cells | ||||
ATRA& the protooncogenes jun and fos (AP-1) | 1-AP-1 plays role in BC cell proliferation and transformation | AP-1 is associated with ATRA-mediated growth inhibition in BC cells | ||
2- Its activity could be inhibited by ATRA |
Table 11.4
Chemopreventive contribution of RARs and RXRs in vivo. (Adapted from Mehdipour et al. 2012)
RARs/RXRs contributions | Isoforms of RARs | Generated by | Conserved modular structure consists | Selective ligand/or as combination | Mode of effect | Leading to |
|---|---|---|---|---|---|---|
1-RARs | Alpha, beta, gamma | Alternative promoters and differential splicing | 1-AF-1 or A/B (ATAF1) Domain | 9-cis-RA | Suppression of carcinogenesis | Reduction in tumor burden |
2- Zinc-finger DBD or C (BD)1 | ||||||
3- CoR or D (HCB3) Domain | ||||||
4- LBD or AF-2 or E (LBTA4) Domain | ||||||
5- Variable F (CT) 5 Domain | ||||||
2-RXR | LGD1069 (targretin) | Suppression of carcinogenesis | Reduction in tumor burden | |||
3- RXR | LGD1069 & tamoxifen | Increased efficacy | Increase in differentiation/decrease in cellular proliferation | |||
4* RXR | ligand LGD1069 & tamoxifen | Increased efficacy | Appropriate response to retinoid therapy |
Cancer prevention is an optimal and end point hope in cancer world, in this regard the list of some environmental factors, their characteristics, and key mechanisms which are involved in the altered epigenetic and genetic make up is provided (Table 11.2). For instance, the dietary methionine, as a vital amino acid, plays a key role in epigenetic which could provide a normal firm for genome-wide DNA methylation patterns through cytosine methylation. In addition, the CpG methylation patterns is important during the early stages of embryon. Therefore, the nutritional behaviors could be translated to the gestations through ancestral line by influencing the epigenetic and genetic composition. So, the fate of individual health, not only, depends on each individual, but on the previous generations as well. Furthermore, research was conducted to investigate an association between folate intake, vitamins B(2), B(6), B(12) and methionine with methylation of promoter region in E-cadherin, p16, and RAR-β(2) genes in the primary BC tumors (Tao et al. 2011). They have stated that intake of these nutrient may be capable to alter promoter methylation in normal or dysplastic breast tissue. In recent publication the multidisciplinary role of epigenetics in cancer prevention as “Nutri- epigenetic” has been, complementary, reviewed (Gerhauser 2013). They have highlighted the influential impact of natural chemopreventive agents on the expression or manner of action of histone modifying enzymes and DNA methyltransferases. The major deregulated events during carcinogenic process by epigenetic alterations include drug metabolism, cell cycle regulation, potential to repair DNA damage/ to induce apoptosis, response to inflammatory stimuli, cell signaling, cell growth control and differentiation. The provided scheme illustrates a complementary view of these machinery (Fig. 11.9). It was also emphasized that epigenetics has influential impact on gene regulation during developmental stages (Gerhauser 2013). It was also highlighted that epigenetic changes occur at early phase of cancer development. In addition they have stated that an interventions with chemopreventive agents, probably, starts early after birth, but I believe that the programming of epigenetic machinery has its root in two complementary territories including heritage and the life style of embryon, i.e. maternal and also ancestral lines including paternal and maternal sides. In addition, a comprehensive insight has been, recently, provided in a book review (Gray SG 2014) in which the basic aspects of epigenetics have been explored.
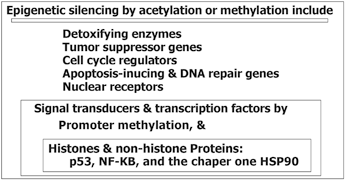
Fig. 11.9
Major deregulated events during carcinogenic process by epigenetic alterations
11.2.1 Stratigical Approaches of Retinoic Acid Receptor β2 Gene
The key role of RAR-β2 gene is highlighted as a tumor suppressing gene which is epigenetically silenced through the course of carcinogenesis. It is shown that the combination of histone deacetylase and DNA methyltransferase are capable to inhibit and reverse the epigenetic silencing capacity of several growth regulatory genes in breast cancer cell lines (Mongan et al. 2005).
Moreover, the chemopreventive impact of retinoids are related to RAR-β2 with its decreased expression in numerous malignant tissues. The first exon expressed in the RAR-β2 transcript is found to be methylated either in ZR-75-1 and SK-BR-3, or in six BC specimens (Widschwendter et al. 2001). They have emphasized on the grading and methylation mode of the lesions by considering the lack of expression of RAR-β2 in grade III and its remarkable carcinogenic impact of this gene on the breast.
The strategic impact of RAR β2 is, partly, due to overexpression of RAR β2 gene, located at chromosome Xq28 and also the transcriptional regulatory mechanisms and immune response (Wallden et al. 2005). The antimetastatic potential of RAR β2 signalling was reported in human breast cancer cells in which following findings were reported:
1.
Overexpression of tumor-cell antigens include cancer testis antigen (CTAG1 and CTAG2), involved in innate immune response i.e., retinoic acid-inducible gene 1/DEAD (Asp-Glu-Ala-Asp) box polypeptide 58 (RIG-I/DDX58), and tumor suppressor functions (i.e., Tyrosinase-Related Protein 1:TYRP1).
2.
Reduced expression by RAR β2 includes CD164 as cell adhesion functions, and FABP6 is involved in metabolic or nutritional processes, and JUN as a transcription factor.
3.
By considering expression profile, they have found diverse capability including “activated and repressed cellular activities in response to overexpression of RARβ2” that have emphasized interesting facts in the metastatic process.
Moreover, epigenetic alterations have key roles in tumorigenesis which has been reported in different types of malignancies, including breast cancer (Sadikovic et al. 2008). Misregulation of specific genes by either genetic or epigenetic alterations are considered as the key facts in cancer development (Sadikovic et al. 2008; Jones et al. 1999).
There are some pitfalls in tumor- formation, progression and management in breast tumors which could be due to epigenetic regulation, combat between epigenetic and genetic territories (which are summarized in Fig. 11.10 (Lo et al. 2008; Novak et al. 2009; Hinshelwood et al. 2008; Tommasi et al. 2009 ). However, the important points are reversibility of epigenetic chromatin modifications (Hendrix et al. 2007), and re-expression of specific targets (Clark et al. 2006). The questions are: (1) Does genomic milieu play key role in reprogramming of tumor suppressor genes? (2) How the required elements and transcriptional activation manage to induce re-expression of silenced territories.
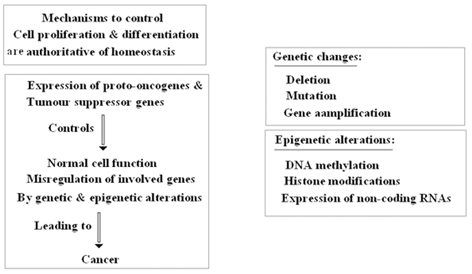
Fig. 11.10
Key role of genetic and epigenetic alterations
Regarding the reprogramming of tumor suppressor gene, the occurance of silencing in the exogenous RARβ promoter of MCF-7 and HCC1954 cell lines was previously, created (Kondo et al. 2008). However, three questions are proposed (1) How is the fate of cancer cell programming through the cell devisions? (2) How the micro- and macro- environmental factors manipulate this process? And (3) Is there any similarity between embryonic territory and post birth genomic and somatic statue?
One of the focal target in cancer prevention is diatry elements, amongst those folate intake is capable to decrease BC risk (Chen et al. 2014). Furthermore and as an example, a recent publication has focused on the essential and required elements by using a biomarker-based validity of food questionnaire (Pirouzpanah et al. 2014a). We have concluded that dietary folate and cobalamin were correlated with the BC-patients’ fasting plasma concentrations. Besides, the dietary methyl group (DMG) have influential impact on the status of hypermethylation of some genes including RARβ2. In this regard, the lower dietary intake of folate or cobalamin, and higher intake of riboflavin or pyridoxine led to an increased incidence of breast tumors development by occuring the promoter methylation of RARβ2 gene (Pirouzpanah et al. 2014b).
However, the associations between DMG intake and the promoter hypermethylation mode of RARβ2 and its expression level in BC patients was recently found by us (unpublished data). This report revealed an association between high dietary riboflavin and pyridoxine intakes with the hypermethylation status of RARβ2. By considering the cumulative nutrient-epigenetic interactions, we have emphasized that the individual who had high dietary vitamin B2 or vitamin B6, are more predisposed to breast tumors with RARβ hypermethylation. In addition, deficiencies of folate and cobalamin were shown to have simultaneous elavatory influence on frequency of tumors characterized with methylated RARβ gene.
11.2.2 Importance of Genetic and Epigenetic Role in Breast Cancer: Rar as a Focal Target
By considering the manner of epigenetic alteration, without change in DNA sequence, i.e., through an opposite direction of genetic changes, the question is whether they cooperate or combat? In this regard, the main fundamental strategies about the key role of genetic and epigenetic alterations is provided (Fig. 11.10). Moreover, in breast tumors there are the following facts at a glance (Allegrucci et al. 2011):
1.
Uncharacteristic epigenetic regulation of cell cycle genes, apoptosis, DNA repair, cell adhesion and signalling leads to tumor formation, manage progression and drug resistance.
2.
In initial developmental stages of breast tumor, epigenetic changes overcome genetic alterations (silencing of CDKN2A (p16INK4A), HOXA and PCDH gene clustering by DNA methylation with over-expression of polycomb proteins BMI-1, EZH2 & SUZ12 during Induced or spontaneously transformation) of human mammary epithelial cells. Methylation of homeobox genes in ductal carcinoma in situ and stage I in breast cancer is also highlighted.
11.2.3 Characteristics Highlights
This item is rather directive and leading paradigm for the future of cancer strategies and managements:
1.
Epigenetic chromatin modifications are reversible which is applicable for attenuation of cancer, like in emryo.
2.
At breast cancer cell lines level: RARβ, CST6, CCND2 (Cyclin D2) were Re-expressed.
3.
Epigenetic modifications of genomic regions mediate silencing of cancer-related genes.
The effect of cellular retinol binding protein I (CrbpI) loss on mammary at RA homeostasis using the Rbp1−/− mouse model was reported (Pierzchalski et al. 2013). It was laso highlighted that type I (CrbpI), is encoded by retinol-binding protein, type 1 (Rbp1). The Rbp assists vitamin A (retinol), acting as chaperone; and revealed to be, epigenetically, silenced in about 25 % of human breast cancers. Such harmonizing process have a key role in proliferation, apoptosis, differentiation, and migration.
Invstigation in vitro showed that the transcription factor RARβ2 is an effective inhibitor of breast cancer cells, proposing the loss of RARβ expression in primary breast cancer. It was reported that expression of the RARβ protein, as the translated product of the human RARβ4 transcript, is elevated in all of five breast tumor cell lines which was related to normal human mammary epithelial cells (Karen et al. 1999).
Loss of expression of RARβ2, is also observed in prostate cancers. Regarding the epigenetic mechanisms and its stable silencing, they have shown the manner of silencing of RARβ2 promoter by harboring two diverse tyrannical chromatin profiles at the same locus, highlighting “the polycomb-mediated epigenetic repression process in cell line prostate cancer (Moison et al. 2013). Morover, the RARβ4 protein is located in the cytoplasm and subnuclear section and its expression is remarkably raised in breast tumor cell lines. They have stated that “RARβ4 functions as a dominant-negative repressor of RAR-mediated growth suppression.”
RARs regulate the genetic network in cancer including breast cancer. It was previously reported that RAR binding act simultaneously with estrogen receptor α (ERα) binding throughout the genome. Such cooperation is reflective of an extensive crosstalk between RA and estrogen signaling through which regulation of the breast cancer-associated genes would be possible (Hua et al. 2009). ERα- and RAR-binding crossroads of two critical nuclear hormone receptor signaling pathways creates a genomic global machinery to balance gene expression.
The characteristics of RA and RARβ2 and its interaction with RARα at cell line level is summarized (Ren et al. 2005):
1.
Resistance to Growth-inhibitory action of Retinoic acid (RA).
2.
Resistance of RA is associated with silencing and hypermethylation of RARβ2 gene.
3.
RARβ2: RA-regulated tumor suppressor gene.
4.
Epigenetically silent RARβ2 relates to lack of RA receptor α (RARα).
5.
RARα regulates RARβ2 transcription by Arbitrating dynamic alteration of RARβ2 chromatin in the presence & absence of RA.
6.
RARβ2 silencing can occur in absence of DNA methylation.
7.
Restoration of RA signal at a silent RARβ2 through RARα leads to RARβ2 reactivation.
8.
RARβ2 silencing and RA resistance lead to an unappropriate RA signal- incorporation at RARβ2 chromatin.
Now, does epigenetic change, such as RARβ2 methylation at diverse tumors’ level is decisive sign to silence this tumor suppressor gene?
In addition, abnormal RARβ2 inactivity may cause repressive epigenetic altertaion at RARβ2, and consequently to RARβ2 silencing and RA resistance (Sirchia et al. 2000, 2002). Besides, normally, dynamic histone changes leads to RARβ2 transcription (in the presence and absence of RA) (Collingwood et al. 1999; Perissi et al. 2004; Dilworth et al. 2001; Xu et al. 1999).
By Bridging and messaging network between RA and RARβ2 chromatin, the following conclusion is provided:
1.
RA resistance may be as the result of an aggravated and extended RARβ2 transcriptional repression; and on the basis of a substandard incorporation of RA signal at RARβ2, with a consequence of diverse factors including genetic, epigenetic, metabolic, micro- and macro- environment.
2.
Abnormal RARβ2 function may be due to the lack of efficient RARα (as the upper regulator of RARβ2 transcription).
3.
RARα has the role of keeping the chromatin of its direct target genes, such as RARβ2, poised for transcription yet inactive.
4.
By RA-RARα binding, the chromatin-mode of the target genes will be renovateed from inactive into active due to the histone alteration, chromatin remodeling, and transcriptional activation.
Besides, in hormone-regulated genes, transcription is normally regulated by dynamic alterations of the chromatin condition in the presence and absence of the relevant hormone (s) (Collingwood et al. 1999; Perissi et al. 2004; Dilworth et al. 2001; Xu et al. 1999). This model may unmask the etiology of aberrant epigenetic silencing in cancer and aging. It was also stated that RARβ2 is commonly epigenetically silenced in RA-resistant cancer cells (Sirchia et al. 2001) and RA-resistant tumors (Sirchia et al. 2002). So, it was hypothesized that this fact is upon to an abnormal status of chromatin-repressive, subsequent lack of essential requirement including RA to facilitate the integration of RA signal at RARβ2. Furthermore, the accumulation of altered histone and DNA level is also presented (Bachman et al. 2003; Stirzaker et al. 2004). Such event in RARβ2, leads to CpG methylation.
Moreover, RA binding to RARα engage coactivator elements with histone acetyltransferase activity which is adequate to switch RARβ2 from a silent to a permissive mode (Perissi et al. 2004). Restoring RA-RARα signaling at RARβ2 is the self reactivation of the RARβ2 receptor (Chiba et al. 1997; Husmann et al. 1991; Sucov et al. 1990). However, Estrogen receptor alpha (ERα) is frequently epigenetically silenced in RARα-negative tumors (Ferguson et al. 1995). Besides, The aberrant hypermethylation of RARβ2 and ERalpha at tumor level has been reported within the Iranian breast cancer patients (Pirouzpanah et al. 2010). As the matter of facts, obesity, duration of estradiol exposure, and smoking are considered as predisposing factors for development of methylation in ERalpha gene. Notably, familial BC was, inversely, correlated with the hypermethylated RARβ2. In addition, plasma folate and vitamin B12 levels were associated inversely with the hypermethylation status of ERalpha gene. These dat suggested that the mode of hypermethylation of specific genes is associated with environmental including lifestyle-related factors.
Interetingly, RARβ2 as a tumor suppressor gene, is involved in carcinogenesis of breast cancer and its silencing is linked to epigenetic chromatin alterations with influential capability on the promoter of RARβ P2. Suppression of chromatin deacetylation at RARβ P2 promoter is occurred due to the DNA methylation (Sirchia et al. 2002). In addition, they could maneuver on the level of histone reacetylation at RARβ P2 either in vivo or in vitro .
For more information, downregulation of the RARβ2 gene is reviewed (Widschwendter et al. 2001). The RARβ2gene as a tumor suppressor gene induces apoptosis and induction of RARβ2 leading to the chemopreventive and therapeutic effects of retinoids. It was highlighted that RARβ2 is reduced or lost, due to the involvement of 5ʹ-region as a cause for loss of expression, through the progression of breat cancer. Interestingly, RARβ2 gene is revealed to be unmethylated either in benign breast tissue or in other normal tissues. In addition, loss of expression of RARβ2 gene was found in prostate cancer. In this regard the directive epigenetic mechanisms to the stable silencing was previously investigated in human prostate tumor cell lines (Moison et al. 2013).
Importantly, by considering gene expression and histopathological features, the methylation status of the RARβ2 gene, and the RARβ2 expression is found to be lower in malignant tissue than in fibroadenoma and normal tissue, but the methylation status was higher in malignant tissue than in normal (Sun et al. 2011). They have highlighted the hypermethylation as an initial event in carcinogenic process of breast.
11.3 About Cancer Stem Cell
Cancer stem cells (CSCs) as a cooperative network interact with many molecular and biological event. The characteristics of CSCs are previously highlighted (Clarke et al. 2006). CSC is a cell within a tumor property which instinctly acquires the capability to self-renew and to establish the heterogeneous lineages of cancer cells. CSC self renew capacity include two manners, (1) “Symmetrical selfrenewing cell division” in which the identical CSCs having self-renewal ability; and (2) “Asymmetrical self-renewing cell division” in which one stem cell and one more differentiated progenitor cell are characterized. CSCs are characterized by their capability to reiterate the generation of an incessantly growing neoplasm. The putative CSCs are also named as ‘‘tumorinitiating cell’’ and ‘‘tumorigenic cell’’. However, symmetrical division of stem cells may form two progenitor cells, leading to depletion of CSCs population or leading to cancer celll death which may be considered as an medication for cancer therapy.
The CSCs have been, initially, isolated in human breast carcinoma (Al-Hajj et al. 2003). The key related characteristics of ductal invasive carcinomas include; (1) Heterogenic intratumoral differentiation of this breast disease, (2) The epithelial to mesenchymal transition (EMT) status, (3) Expression of TWIST1, as a repressor of an EMT-inducing transcriptional factor in invasive lobular breast cancer, (4) Disuniting of invasive tumor cells in EMT-like condition, (5) different pathways are involved in formation of cancer stem cells, and (6) Distribution of associated tumor cell, are all correlated with the malignant progression leading to a poor clinical prognosis (Yang et al. 2004; Fujita et al. 2003; Xue et al. 2003; Blanco et al. 2002).
Due to these characteristics breast cancer is a focal territory in which two paradigms including cancer stem cells and EMT of cancer stem cell migration is remarkably highlighted.
Moreover, the intestinal gastric cancer, pancreatic cancer, and squamous cell carcinomas are also the examples for this category in which the intra-tumor heterogeneity and EMT could be detected (Nakajima et al. 2004; Rosivatz et al. 2002; McAlhany et al. 2004).
Three main fscts about stem cells in solid tumors are highlighted as followings:
1.
Are less accessible.
2.
Lack of the appropriate functional assay for tracing and quantifying normal stem cells from different organs.
3.
Limitation in isolation of stem cells by the cell surface markers in human.
Regarding the genetic and epigenetic signatures of ‘‘Stemness’’ Clarke and his colleagues (Clarke et al. 2006) have stated very important considerations as:
1.
“To identify true signatures, pure populations are necessary.” This is especially true for cells expected to be rare, such as cancer stem cells, whose expression signature would be swamped by the majority of nonstem cells in a whole tumor sample.
2.
“Even after a cancer stem cell signature from a particular type of tumor is identified, one cannot assume that a given signature is useful for identifying cancer stem cells in a different tumor type unless validated by a functional assay (such as an in vivo self-renewal assay as it is the most definitive at this point in time).”
However, as a complementary information the essential renewal assay in vivo is not adequate to be translated in human and more specific definition is required for evaluating the quantitative values of stemness in human cancers. Such attempts require the follow-up strategy during different stages of cancer patients. Although microarray and genomewide technologies are applied to unmask tendency in genetic and epigenetic for CSCs, but specific Cell Based Strategy (CBS) would, appropriately, solve the heterogeneity insight of diverse tumors.
Labelling a cell as stem cell and cancer stem cell is a rather difficult aim. There are some key questions; (1) Where is the stem cell originated from? Is it derived from other stem cell populations? Or (2) is it derived from an initially normal cell or (3) Or derived from cancer cell?
Now lets to characterize the capability of in vitro assay:
Reliability of an in vitro assay to define a cell as “stemness” and cancer stem cell:
1.
Reflects the characteristic of an early stage.
2.
The results require the complementary/confirmative/validitative (CCV) at vivo level.
3.
The complementary functional assay (s) is required.
4.
Gene activity profiles, gene expression signatures and/or cell surface marker are required.
5.
There are some pitfalls in cell analyzing due to heterogenetic character of cancer cells and the techniques through which the results reflect a global insight and not individualized characteristics. To support this matter, an example is the expression assay by real time PCR which is considered as a global insight. Th spectrum for categorizing the cancer cell population is diverse and an exact grouping profile is essential to narrow and escape the subgrouping problem. Relative signature of cellular and molecular targets is required for providing more reliable definition for certain population of cancer cells.
6.
There is a double edged sward including inactivation or activation of target gene (s) to reduce or produce stem cells respectively. This would support the therapeutic machinery in clinical application. Besides, what does matter would be resistance of cancer cells in target based therapy.
As the matter of fact and by considering the epithelial to mesenchymal transition/mesenchymal to epithelial transition (EMT/MET) processes, there are a triangle in cancer initiation including programming/reprogramming/cancer progression. Furthermore, stem cells are associated with embryonic stem (ES) cells or induced pluripotent stem cells (iPSCs), immortalized mammary epithelial cells that have undergone epithelial to EMT transition which is the reverse manner of the MET process (Mani et al. 2008; Morel et al. 2008). There is also a cooperation between two capacities including EMT and stemness through cancer progression, and separation of cells from the primry tumor that facilitate migration and metastasis. Interestingly, such evocative process of EMT is similar to the embryonic tissue development which is the state of art and fascinating journey of cells from far away departure station to diverse destinations (Thiery et al. 2009). Also, CSCs are capable to kernel new neoplasms to self-renew and lead to generate non-stem differentiated cells. CSCs is not apparently pluripotent which differentiate them from iPSCs; due to this point, cancer cells do not produce any cell type; instead may be considered as relapse of the original primary tumor cells occurs through metastatic process (Gupta et al. 2009; Polyak et al. 2009). However, the machinery of stem cell and invasion is rather a core item in cancer metastasis. In this regard, it worths to refer to the recent work which was focused on the “mouse haematopoietic stem cell regulator Latexin (LXN)” (Oldridge et al. 2013). This target was considerd as a unique “homologue of the retinoic acid receptor responder 1 (RARRES1) gene”. The co-expression of either RARRES1 or LXN was suppressed by DNA methylation in prostate cancer (PC) cell lines and furthermore inhibition of RARRES1 and LXN led to increase the invasive capability of primary PC cell line.
Additionally, it is recently reported that phosphorylation of the RARγ2 play a key role for the neuronal differentiation embryonic stem cells in mouse (Al Tanoury et al. 2014).
11.3.1 Modelling Cancer Stem Cell
As a matter of fact the dialougue between genetics, epigenetics and the relevant diversities are the partial reason for heterogeneity of cancer cells.
Now, by considering the common and uncommon characteristics in variety of cancers, important points are adressed as; (1) Is there any cancer model for specific cancer? (2) However, it was stated that the cause of the pyramid growth and progression in cancers has its roots in “small subpopulations of cancer stem cells” (Reya et al. 2001; Dick 2008). Also, do cancer stem cells experience relatively irreversible epigenetic alteration (s) to form different population of nontumorigenic cancer cells? However, There is battle between diverse subpopulation of cells including tumorigenic and nontumorigenic which are differentiatable from each other by limited available expression profiling assays which was stated to be, morphologically, unclear (Al-Hajj et al. 2003).
Furthermore, the wonder is that ‘Tumorigenic cancer stem cells may form diverse Nontumorigenic cells.’ And ‘How could we differentiate tumorigenic cells from nontumorigenic cells? Is it due the epigenetic event? Is it as a result of either epigenetic or genetic events?’
There are few publication on cancer stem cell model in limited number of human cancers and in mouse (Kelly et al. 2007; Williams et al. 2007; Quintana et al. 2008; Quintana et al. 2008), but still the evolutionary/clonal model by Nowell seems to be more precisely (Nowell 1976). The focal points of this model include the heterogenic faetur of cancer stem cells (CSC), hetrogenic/homogenic feature of clones, diverse tumorigenic capability of CSC; and organization of tumor. In addition this model is characterized with diverse nature of tumorigenic and nontumorigenic cells which are affected by epigenetic in CSCs, and by genetic or epigenetic in clonal model. Furthermore, involvement of diversity in epigenetic and genetic is an explanation for therapeutic resistance in various cancers among tumorigenic cancer (Nowell 1976).
Diversity in epigenetic event between cancer stem cells and their progeny could verify the clinical manners of specific cancers, but clonal evolution has a key role in all neoplasms.
When the cancers are classically developed into epigenetically differentiable populations of tumorigenic and nontumorigenic cancer cells, the occurrence of clonal evolution in the CSCs, plays a key gate role (Barabe et al. 2007).
Some facts and questions in cancer cell heterogeneity include:
1.
Tumorigenic capability due to epigenetic and/or genetic.
2.
Does cancer cells differentiate to a nontumorigenic status?
3.
5.
By considering the clonal evolutionary model, genetic heterogeneity in cancer cells could lead to heterogenic feature, cellular function, and finally response to the therapeutic protocols.
6.
Epigenetic diversity governs the ongoing events by leading to more complementary heterogenic behavior.
7.
In cancer stem cell model, the classical structure of tumorigenic and nontumorigenic cells is due to the native epigenetic diversity within the cancer cell populations (Reya et al. 2001; Dick 2008). In this regard the provided image is indicative of such diversity in stem cells (Fig. 11.11).
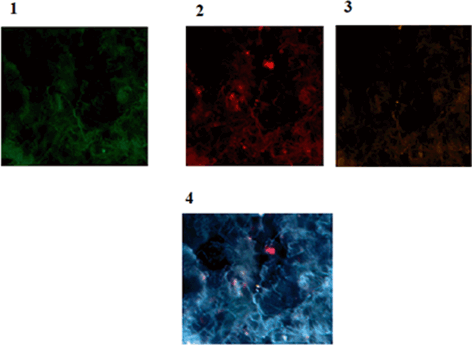
Fig. 11.11
Protein expression of p53, CD44 and CD24 in a patient affected with breast cancer. 1 Tumor of breast conjugated with FITC reflecting low expression of p53, 2 Same cells conjugated with Pe-Cy5 reflecting diverse expression of CD44 including low, medium and high (In limited cells), 3 Same cells conjugated with R-Pe reflecting lack of expression of CD24, accompanied by an isolated cell with low expression, 4 Co-expression of p53, CD44 and CD24 presenting the presence of limited cancer stem cell cooperating with CD44 Magnification:x200. From: P.Mehdipour’s archive
8.
Does clonal evolution leads to heterogeneity and forms a tumor consisting of a unique structure of tumor cells which have lots in common?
9.
Does misregulated self-renewal pathways cause differentiable epigenetically tumorigenic and nontumorigenic cells in all cancers?
11.4 An Interaction Insight in Epigenetic by Focusing on the ER, PR, HER2 Triangle Targets
One of the serious concern in breast cancer is the possible developmental capacity of malignancy in premalignant breast neoplasm. Such process by considering the status of DNA methylation including Methylated-IN-Tumor (MINT)17, MINT31, RARβ2 and RASSF1A genomic markers have been studied through benign, premalignant and malignant status of breast cancer (van Hoesel et al. 2013). They have found DNA hypermethylation at early stage of BC development with diverse degree of tumor during the cancer progression.
Related posts:
 Reduced Risk of Cancer in Schizophrenia, a Bridge Toward Etio-Pathology and Therapy of Both Diseases
Reduced Risk of Cancer in Schizophrenia, a Bridge Toward Etio-Pathology and Therapy of Both Diseases
 Biodynamic Phenotypic and Epigenetics Changes of Circulating Tumor Cells: Their Application in Cancer Prognosis and Treatment
Biodynamic Phenotypic and Epigenetics Changes of Circulating Tumor Cells: Their Application in Cancer Prognosis and Treatment
 An Introduction to Impact of Bio-Resonance Technology in Genetics and Epigenetics
An Introduction to Impact of Bio-Resonance Technology in Genetics and Epigenetics
 Epigenetics of Thyroid Cancer
Epigenetics of Thyroid Cancer
 Malignant Rhabdoid Tumor: Epigenetic Mechanism of Tumorogenesis
Malignant Rhabdoid Tumor: Epigenetic Mechanism of Tumorogenesis
 Sentinel Gene Within Cell Territory and Molecular Platforms in Cancer: Methylation Diversity of p53 Gene in Brain Tumors
Sentinel Gene Within Cell Territory and Molecular Platforms in Cancer: Methylation Diversity of p53 Gene in Brain Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


