Eosinophilic Neoplasms and Hypereosinophilic Syndrome
Eosinophilic Neoplasms and Hypereosinophilic Syndrome
Huong (Marie) Nguyen
Jason Gotlib
HISTORICAL BACKGROUND
Paul Ehrlich’s use of aniline dyes facilitated identification of the eosinophil as a unique leukocyte in 1879.1 Reactive eosinophilias and eosinophilia-associated leukemias were recognized in the early to mid-twentieth century. However, a nomenclature for unexplained eosinophilias was not formulated until 1968 when Hardy and Anderson originated the term hypereosinophilic syndrome (HES).2 In 1975, Chusid and colleagues generated diagnostic criteria for (idiopathic) hypereosinophilic syndrome based on a small case series of patients: (1) persistent eosinophilia of 1.5 × 109/L for more than 6 months; (2) lack of evidence for parasitic, allergic, or other known causes of eosinophilia; and (3) signs and symptoms of organ involvement.3 In the last 20 years, the evaluation of eosinophilic disorders has advanced from its descriptive roots to a more comprehensive approach which relies on a combination of histopathology, cytogenetics, molecular genetics, and immunophenotyping. Table 84.1 lists recent modern nosologic and clinicopathologic landmarks which reflect this evolution in the diagnosis and classification of eosinophilic disorders.
DEFINITION OF EOSINOPHILIA
Although minor differences may exist between laboratories, the upper limit of normal for the percent peripheral blood eosinophils is 5%, with a corresponding absolute eosinophil count (AEC) of approximately 0.5 × 109/L (500/mm3).4,5 The severity of hypereosinophilia (HE) has been stratified into mild, moderate, or severe (with AECs between the upper range of normal to 1.5 × 109/L (1,500/mm3), 1.5 to 5.0 × 109/L (1,500 to 5,000/mm3), or >5.0 × 109/L (>5,000/mm3), respectively). Alternatively, the singular term hypereosinophilia has been used to denote an AEC of >1.5 × 109/L (>1,500/mm3).
TABLE 84.1 MODERN CLASSIFICATION AND CLINICOPATHOLOGIC LANDMARKS IN EOSINOPHILIC DISORDERS
Year
Event
1968
Term “hypereosinophilic syndrome” coined by Hardy and Anderson
1975
Diagnostic criteria for HES established by Chusid and colleagues
1994
Characterization of the first PDGFRB rearrangement (ETV6-PDGFRB), t(5;12)(q31-33;p13)
1994
First description of lymphocyte-variant hypereosinophilia
1998
Identification of rearrangement of the FGFR1 gene as the basis for the 8p11 syndrome
2001
WHO diagnostic criteria for HES and CEL
2001-2002
Successful empiric treatment of HES patients with imatinib
2002
Characterization of the first PDGFRA rearrangement (BCR-PDGFRA), t(4;22)(q12q11)
2003
Identification of the FIP1L1-PDGFRα fusion as the therapeutic target of imatinib
2003
Identification of FIP1L1-PDGFRA using the surrogate test “FISH for the CHIC2 deletion”
2008
Revised WHO criteria include the category of “Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1”
FISH, fluorescent in situ hybridization; HES, idiopathic hypereosinophilic syndrome; PDGFRA; platelet-derived growth factor receptor alpha; PDGFRB, platelet-derived growth factor receptor beta; WHO, World Health Organization.
EPIDEMIOLOGY
HES and chronic eosinophilic leukemia (CEL) are very uncommon disorders. Data obtained from the Surveillance, Epidemiology, and End Results (SEER) program during the years 2001 to 2005 revealed an age-adjusted incidence rate of 0.036 per 100,000.6 Eosinophilias with recurrent genetic abnormalities (PDGFRA/B, FGFR1) are yet even more rare, representing approximately 10% cases of initially unexplained eosinophilias in developed countries.7 SEER identified a peak age range at diagnosis of 65 to 74 years, with cases uncommonly diagnosed in infants and children. The male to female ratio of HES/CEL is approximately 1.5:1, but FIP1L1-PDGFRA-positive disease is almost exclusively diagnosed in men, a phenomenon whose biologic basis remains unknown.6,7
EOSINOPHIL PHYSIOLOGY
Eosinophil-committed hematopoietic progenitors are identified within the CD34+ progenitor population as interleukin-5 receptor (IL5R)+/CCR3+ cells.8,9 Their production is tightly regulated by a network of eosinophilopoietic cytokines. IL-5, interleukin-3 (IL-3), and granulocyte-macrophage colony-stimulating factor are the most relevant to eosinophil physiology, and are primarily responsible for the commitment, proliferation, and differentiation of hematopoietic progenitors into the eosinophil lineage.4,10 These factors are elaborated by several cell types including T-lymphocytes, mast cells, and stromal cells, and bind to their respective receptors on eosinophils.11 Migration of eosinophils from the circulation to tissues is mediated via interactions between endothelial cell adhesion molecules such as ICAM-1 and VCAM1 and eosinophil surface integrins (e.g., the α1 and β2 integrins VLA-4 and CD18, respectively).12 Eosinophil migration is influenced by several potent eosinophil chemoattractants including leukotriene β4, complement fragment 5a, platelet activating factor, RANTES, and eotaxins, which are ligands for the eosinophil receptor CCR3.13,14 and 15 Although eosinophils serve as an essential component of the immune system’s normal homeostatic function, including defense against infection and recruitment in response to allergy and inflammation, the potential for collateral tissue injury exists due to mediators released from eosinophils undergoing marked or persistent activation. These biologically active molecules are released from intracellular granule compartments and include preformed substances such as major basic protein, eosinophil peroxidase, eosinophil cationic protein, and eosinophilderived neurotoxin.4 Eosinophils also contain hydrolytic enzymes such as acid phosphatase, catalase, arylsulfatase, and newly synthesized mediators such as hydrogen peroxide that can contribute to organ damage.4
MODERN CLASSIFICATION
The most recent iteration of the World Health Organization (WHO) classification of hematolymphoid neoplasms incorporates eosinophilic disorders among the umbrella of myeloid neoplasms (Table 84.2). The WHO classification recognized the unique molecular basis of a subset of certain eosinophilic disorders by establishing the major category entitled, “Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of platelet-derived growth factor receptor alpha (PDGFRA), platelet-derived growth factor receptor beta (PDGFRB), or fibroblast growth factor receptor 1 (FGFR1)” (Table 84.3).16 The diagnostic entity “chronic eosinophilic leukemia not otherwise specified (CEL-NOS)” is one of eight diseases included within another major WHO category, “Myeloproliferative Neoplasms (MPNs)” (Table 84.3).17
CEL-NOS is defined by the absence of the Philadelphia chromosome or a rearrangement involving PDGFRA/B or FGFR1, and the exclusion of other WHO-defined acute or chronic primary marrow neoplasms associated with eosinophilia, e.g., acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), MPNs such as chronic myelogenous leukemia (CML) or systemic mastocytosis (SM), and overlap MDS/MPNs. It is also defined by an increase in blood or marrow blasts (<20%), and/or nonspecific cytogenetic abnormalities. The diagnostic criteria of HES formulated by Chusid and colleagues is recapitulated by the WHO, but the original requirement that eosinophilia persist for more than 6 months is no longer consistently embraced. This relates to the faster-paced and more sophisticated evaluation of eosinophilia that is now feasible, and the recognition that early treatment can be critical to mitigating eosinophilia-mediated organ damage. The nosological distinction between “idiopathic hypereosinophilia” and “idiopathic HES” reflects the requirement that organ damage be present in the latter. “Lymphocyte-variant hypereosinophilia” is a provisional diagnosis that is referred to in the WHO classification and should be considered before settling on the diagnosis of idiopathic hypereosinophilia or HES, which are diagnoses of exclusion.17
TABLE 84.2 2008 WORLD HEALTH ORGANIZATION CLASSIFICATION OF MYELOID MALIGNANCIES
Acute myeloid leukemia and related disorders
MPNs
Chronic myelogenous leukemia, BCR-ABL1 positive
Chronic neutrophilic leukemia
Polycythemia vera
Primary myelofibrosis
Essential thrombocythemia
Chronic eosinophilic leukemia, not otherwise specified
Diagnostic criteria of MPN associated with ETV-6-PDGFRB fusion gene or other rearrangement of PDGFRB
An MPN, often with prominent eosinophilia and sometimes with neutrophilia and monocytosis and
Presence of t(5;12)(q31˜q33;p13), or a variant translocationc, or demonstration of an ETV6-PDGFRB fusion gene or rearrangement of PDGFRB
Diagnostic criteria of MPN or acute leukemia associated with FGFR1 gene rearrangement
An MPN with prominent eosinophilia and sometimes with neutrophilia or monocytosis or AML or precursor T-cell or precursor B-cell lymphoblastic leukemia/lymphoma (usually associated with peripheral blood or bone marrow eosinophilia) and
Presence of t(8;13)(p11;q12) or a variant translocation leading to FGFR1 rearrangement demonstrated in myeloid cells, lymphoblasts, or both
Chronic Eosinophilic Leukemia-NOS
Eosinophilia (eosinophil count > 1.5 × 109/L)
No Ph chromosome or BCR-ABL fusion gene or other MPNs (PV, ET, PMF, systemic mastocytosis) or MDS/MPN (CMML or atypical CML)
No t(5;12)(q31˜q35;p13) or other rearrangement of PDGFRB
No FIP1L1-PDGFRA fusion gene or other rearrangement of PDGFRA
No rearrangement of FGFR1
Blast cell count in the peripheral blood and bone marrow is less than 20% and there is no inv(16)(p13q22) or t(16;16)(p13q22) or other feature diagnostic of AML
There is a clonal cytogenetic or molecular genetic abnormality, or blast cells are more than 2% in the peripheral blood or more than 5% in the bone marrow
WHO-defined myeloid malignancies associated with eosinophilia (e.g., MDS, MPNs, MDS/MPNs, or AML)
Eosinophilia-associated MPNs or AML/ALL with rearrangements of PDGFRA, PDGFRB, or FGFR1
Absolute eosinophil count of >1,500/mm3 must persist for at least 6 mo and tissue damage must be present. If there is no tissue damage, idiopathic hypereosinophilia is the preferred diagnosis
a Patients presenting with AML or lymphoblastic leukemia/lymphoma with eosinophilia and an FIP1L1-PDGFRA fusion gene are also assigned to this category.
b If appropriate molecular analysis is not available, this diagnosis should be suspected if there is a Ph-negative MPN with the hematologic features of chronic eosinophilic leukemia associated with splenomegaly, a marked elevation of serum vitamin B12, elevation of serum tryptase, and increased bone marrow mast cells.
c Because t(5;12)(q31˜q33;p12) does not always lead to an ETV6-PDGFRB fusion gene, molecular confirmation is highly desirable. If molecular analysis is not available, this diagnosis should be suspected if there is a Ph-negative MPN associated with eosinophilia and with a translocation with a 5q31-33 breakpoint.
In 2011, members from the Working Conference on Eosinophil Disorders and Syndromes proposed a new terminology for eosinophilic syndromes.18 The panel recommended the higher level term “Hypereosinophilia (HE)” for persistent and marked eosinophilia (AEC > 1.5 × 109/L). In turn, HE subtypes were divided into a hereditary (familial) variant (HEFA), HE of undetermined significance (HEUS), primary (clonal/neoplastic) HE produced by clonal/neoplastic eosinophils (HEN), and secondary (reactive) HE (HER). HEUS was introduced as a novel term in lieu of “idiopathic hypereosinophilia.” Any HE (not just idiopathic) associated with organ damage is referred to as “HES” with specific variants designated by subscripts (e.g., HESUS, HESN, and HESR). Additional recommendations advanced by the consensus panel are summarized in their report.18
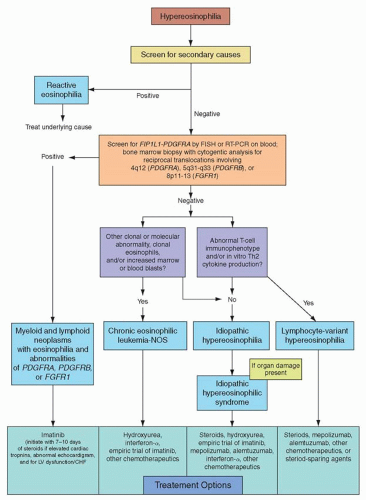
FIGURE 84.1 Algorithm for the diagnosis and treatment of eosinophilic disorders.
DIAGNOSTIC EVALUATION FOR HYPEREOSINOPHILIA
In addition to a hematology consultation, a multi-disciplinary evaluation including allergy/immunology, dermatology, and infectious diseases/tropical medicine may be required to identify the etiology of HE. The workup of HE (Fig. 84.1) should commence with a thorough evaluation of reactive causes, with particular attention to comorbid health conditions, medications, and travel history/environmental exposures. Helminth infections are the most common cause of HE in developing countries, and hypersensitivity/atopy is the most frequent etiology in industrialized nations. In addition to infection and hypersensitivity conditions, other secondary causes of HE include connective tissue disorders (Churg-Strauss syndrome), granulomatosis with polyangiitis (Wegener’s, systemic lupus erythematosus), solid or hematologic malignancies, metabolic disorders (e.g., adrenal insufficiency), eosinophilic pulmonary diseases, and rare entities such as familial eosinophilia,19 hyper-immunoglobulin (Ig)E syndrome, Omenn syndrome, episodic angioedema and eosinophilia (Gleich’s syndrome), and eosinophilia-myalgia syndrome (possibly related to tryptophan ingestion and the historical epidemic of toxic-oil syndrome).20,21
In addition to a complete blood count with a white blood cell count (WBC) differential to measure the severity of eosinophilia, the initial laboratory workup for reactive causes of HE typically includes stool culture and serial ova and parasite testing, and sometimes serologic testing for specific parasites (e.g., strongyloides antibody). Testing for specific bacteria, fungi, or viruses may also be warranted in selected cases. The use of chest x-ray, electrocardiogram and/or echocardiography, and computed tomography scan of the chest or abdomen/pelvis are similarly guided by the patient’s symptoms and physical exam findings. Imaging studies may help discern the basis for eosinophilia, establish the presence or severity of organ damage, and guide the need for tissue biopsy. For eosinophilic lung diseases, pulmonary function testing, bronchoscopy, and serologic tests (e.g., aspergillus IgE to evaluate for allergic bronchopulmonary aspergillosis) may be obtained to further characterize lung involvement.
If secondary causes of eosinophilia are excluded, the work-up should proceed to the evaluation of a primary bone marrow disorder. Bone marrow aspirate and trephine biopsy with cytogenetics and immunophenotyping (with relevant iron and reticulin/trichrome stains) are useful to elucidate WHO-defined clonal myeloid neoplasms associated with eosinophilia such as acute myeloid leukemia, specifically AML with inv(16)(p13q22) or t(16;16)(p13;q22), CML or other MPNs (e.g., SM), and overlap MDS/MPN disorders such as chronic myelomonocytic leukemia (CMML).17
Myeloid malignancies primarily defined by eosinophilia include the aforementioned CEL-NOS and the category of PDGFRA/B or FGFR1-rearranged neoplasms. Diagnostic assessment of primary eosinophilia should begin with screening of the peripheral blood for the FIP1L1-PDGFRA gene fusion by interphase or metaphase fluorescence in situ hybridization (FISH).22 Less commonly, reverse-transcriptase polymerase chain reaction (PCR) is used to detect the fusion. FISH probes that hybridize between the FIP1L1 and PDGFRA loci detect the 800-kb interstitial deletion on chromosome 4q12 that results in gene fusion. Because the CHIC2 gene is located in this deleted genetic segment, the test has been referred to as “FISH for the CHIC2 deletion.”23 If FIP1L1-PDGFRA screening is not available, serum tryptase has been used as a surrogate marker for FIP1L1-PDGFRA-positive disease, since increased levels have been associated with the fusion or myeloproliferative forms of HE.24 In contrast to FIP1L1-PDGFRA, which is not detectable by standard karyotyping, rearrangements involving PDGFRB and FGFR1 can be inferred by cytogenetic analysis, represented by the breakpoints 5q31-33 (PDGFRB) and 8p11-12 (FGFR1), respectively.16 Reciprocal chromosomal translocations involving the breakpoints 9p24 or 13q12 may relate to JAK2 (e.g., PCM1-JAK2) or FLT3 (e.g., ETV6-FLT3) fusions, respectively, and have been associated with eosinophilia.25,26 Knowledge of these breakpoints is critical because targeted therapy may have clinical efficacy against these constitutively activated tyrosine kinases. Bone marrow biopsy studies as highlighted above should be performed to complement peripheral blood analyses.
If both secondary and primary causes of eosinophilias are excluded, lymphocyte-variant HE should be considered next in the diagnostic algorithm.27 Patients with lymphocyte-variant HE often have cutaneous signs and symptoms as the primary disease manifestation. Although patients’ skin disease can be symptomatic, the natural history of this condition is typically indolent, with rare patients progressing to T-cell lymphoma or Sézary syndrome. Progression has been associated with acquisition of cytogenetic abnormalities (e.g., partial 6q and 10p deletions, trisomy 7) in T cells and with proliferation of lymphocytes with the CD3–CD4+ phenotype.28,29,30,31,31 This syndrome represents a mixture of clonal and reactive processes resulting in the expansion of a clone of T-lymphocytes that produce cytokines that drive eosinophilia.27,32,33 Although these laboratory findings constitute basic elements of this syndrome, neither the WHO nor other consensus panels have established specific diagnostic criteria for this condition.16,17 and 18 The finding of isolated T-cell clonality by PCR without T-cell immunophenotypic abnormalities or demonstration of Th2 cytokine production is not adequate to make a diagnosis of this variant.32 In an analysis of patients diagnosed with HES, 18/42 (43%) subjects exhibited a clonal T-cell receptor gene rearrangement by PCR.34 However, the biologic relevance of such clonal T-cell populations to eosinophilia was not established.34 Therefore, whether such patients should still be referred to as HES, or as lymphocyte-variant HE, remains a matter of debate. Laboratory features that suggest this disorder are shown in Table 84.4.
BIOLOGY OF FIP1L1-PDGFRA
FIP1L1-PDGFRA can promote the proliferation and survival of eosinophils through the activation of several signaling pathways such as phosphoinositol 3-kinase (PI3 kinase), ERK1/2, and STAT5.22,35 The exact mechanism by which FIP1L1-PDGFRα preferentially affects eosinophils remains unclear. Recently, it was found that in vitro inhibition of JAK2 in the FIP1L1-PDGFRA-positive EOL-1 cell line, primary FIP1L1-PDGFRA-positive cells, and T674I FIP1L1-PDGFRA-imatinib-resistant cells, by either JAK2-specific short interfering RNA or the tryphostin derivative AG490 (a JAK inhibitor), significantly reduced cellular proliferation and induced cellular apoptosis.36 JAK2 inhibition also reduced PI3 kinase, AKT, and nuclear factor kappa B activity in a dose-dependent manner, and suppressed expression levels of c-myc and survivin.36 The results suggest that JAK2 is activated by FIP1L1-PDGFRα and is required for cellular proliferation, possibly through induction of c-myc and survivin.
TABLE 84.4 LABORATORY FINDINGS IN LYMPHOCYTE-VARIANT HYPEREOSINOPHILIA
Cytokine-producing abnormal T-lymphocyte population with aberrant immunophenotype
Double-negative, immature T cells (e.g., CD3+CD4–CD8–), or
Absence of CD3 (e.g., CD3–CD4+), a normal component of the T-cell receptor, or
Elevated CD5 expression on CD3–CD4+ cells, or
Loss of surface CD7 and/or expression of CD27
Reactive eosinophilia in response to T-lymphocyte secretion of eosinophilopoietic cytokines
Elevated serum IgE levels
Lymphocyte production of cytokines (e.g., IL-5, IL-4, IL-13) suggesting Th2 cytokine profile
Elevated production of TARC, a chemokine in Th2-mediated diseases
IL, interleukin; TARC, thymus and activation-regulated chemokine; Th2, T cells with helper type 2.
In one murine model, expression of the FIP1L1-PDGFRA fusion in bone marrow cells was not sufficient to cause eosinophilia, but only a general myeloproliferative disease.37 However, in another murine model, expression of FIP1L1-PDGFRA together with overexpression of IL-5, the most potent eosinophilopoietic cytokine, mimicked more typical features of HES, such as tissue infiltration of eosinophils.38 Polymorphic variation at the IL-5 receptor-a (IL5RA) gene revealed an association between a single nucleotide polymorphism in the 5′ untranslated region of IL5RA and the eosinophil count/presence of tissue infiltration in FIP1L1-PDGFRA-positive patients.39 These data suggest that FIP1L1-PDGFRA alone is not sufficient to explain the development of HES/CEL, and that additional factors such as IL-5 signaling may also be implicated in the disease phenotype.
The structure of the FIP1L1-PDGFRa fusion protein is similar to the structure of the ETV6-PDGFRα, ZNF198-FGFR1, and BCR-ABL1 proteins, for which homotypic oligomerization mediated by domains within ETV6, ZNF198, or BCR has been reported.40,41 and 42 Oligomerization of the corresponding fusion proteins leads to activation of the tyrosine kinase domains, which in turn activate downstream signaling pathways regulating cell proliferation and survival. In contrast, interruption of the juxtamembrane of PDGFRα, either due to mutations or duplications, causes constitutive activation of kinase activity.43,44 This mechanism occurs with internal tandem duplications in FLT3 and mutations of KIT in AML or gastrointestinal stromal tumors.45,46 Fusion of FIP1L1 to the PDGFRa protein yields a constitutively active tyrosine kinase only if the juxtamembrane domain of PDGFRα is partially or completely removed.47 The different breakpoints within the PDGFRA gene are tightly clustered, resulting in the removal of part of the juxtamembrane domain and activation of the kinase domain.48 In contrast, with many PDGFRB fusions, the juxtamembrane is completely intact, and activation of PDGFRB kinase activity is obtained through oligomerization mediated by the fusion partner.
Only gold members can continue reading. Log In or Register to continue
 Clinical Flow Cytometry
Clinical Flow Cytometry



