Fig. 13.1
(a) Nitric oxide (NO) is produced from l-arginine and molecular oxygen (O2) by endothelial nitric oxide synthase (eNOS) in a tightly “coupled” process involving tetrahydrobiopterin (BH4) and NADPH. (b) In diabetes, increased redox imbalance (due to increased NADH/NADPH) and decreased availability of BH4 (due to oxidation) leads to “uncoupling” of NO production. This results in transfer of electrons to O2 to form superoxide (O2 •−). Superoxide in turn reacts with and consumes NO, forming the oxidant species peroxynitrite (OONO−). Hence, oxidative stress and endothelial dysfunction are further increased [14]. Adapted from Katusic ZS [15]
In Vivo Measurement of Endothelial Function
Endothelial function may be measured indirectly in the peripheral circulation by assessing the vasodilatory responses of conduit and resistance arteries to stimuli that increase NO release (Table 13.1) [3, 13, 16]. In the brachial artery, shear stress is generated by hyperemia following an induced period of local ischemia, and flow-mediated dilatation (FMD) is measured using high-resolution ultrasonography or even magnetic resonance imaging [17, 18]. Blood flow changes in the forearm microcirculation following hyperemia, or intra-arterial infusion of muscarinic receptor agonists such as acetylcholine, can be measured using venous occlusion strain-gauge plethysmography [19, 20]. An emerging non-invasive clinical tool to assess ED is digital peripheral arterial tonometry (PAT) (Endo-PAT, Itamar Medical) [21, 22].
Table 13.1
Techniques and methods for assessing endothelial function in humans
Coronary circulation | Peripheral circulation | Circulating biomarkers |
|---|---|---|
QC Angiography | Ultrasonography: FMD | ADMA, NO |
PE Tomography | Plethysmography: FABF | ET-1 |
Endo-PAT | hs-CRP | |
vWF | ||
Vasodilatory Stimuli | PAI-1 | |
Acetylcholine | ICAM, VCAM | |
Shear Stress | Selectins | |
Nitrates | EP Cells | |
NOS inhibitors | EMP | |
Endothelial function in coronary arteries may also be assessed, in response to pharmacological agonist or shear stress stimuli, using quantitative angiography to measure vessel diameter changes [23]. Non-invasive methods, such as positron emission tomography, may also be undertaken, but are costly [13].
Circulating biomarkers may be measured as indirect indices of endothelial cell damage, activation and inflammation (Table 13.1) [24–30]. Impaired mobilization or depletion of endothelial progenitor cells derived from bone marrow are involved in the pathogenesis of ED, and their circulating levels can also be used as a marker of ED [31–34]. Recently, a relationship between progenitor cells and cell-derived microparticles has been demonstrated [33]. Microparticles (MP) are small membrane-shed vesicles derived from cell surfaces under conditions of cellular activation or injury/apoptosis [33, 35]. Thus, endothelial-derived microparticles (EMP) may be potential markers of ED [33, 35]. Vascular extracellular superoxide dismutase (ecSOD) activity, the major antioxidant enzyme system of the vessel wall, was substantially reduced in patients with CAD and closely associated with NO-mediated vasodilation, suggesting that reduced ecSOD activity contributes to the reduced bioavailability of NO [36]. However, measurement of ecSOD requires the intravenous injection of heparin; therefore, its utility as a surrogate marker of ED in the clinical setting is less practicable.
Endothelial Dysfunction
Endothelial Dysfunction: Uncoupling of eNOS
Endothelial dysfunction reflects an imbalance between release of vasodilator and vasoconstrictor endothelial-derived factors. A decrease in the bioavailability of NO, involves either a decrease in NO synthesis or inactivation of NO due to increased endothelial production of reactive oxygen species (ROS) [37]. With increased oxidative stress, tetrahydrobiopterin (BH4), a cofactor that tightly regulates NO production, is oxidized resulting in the uncoupling of eNOS and reduced NO production [14]. Elevated levels of asymmetric dimethylarginine (ADMA), an endogenous inhibitor of eNOS through competition with l-arginine, may further reduce NO production [13]. This perpetuates a cycle of vascular oxidative stress through the transfer of electrons to molecular oxygen, forming oxidant species such as superoxide and peroxynitrite, which further consumes NO and increases oxidative stress (Fig. 13.1b) [14, 38].
Predictive Value of Endothelial Dysfunction
Several studies in diverse groups of subjects have shown that ED measured by the aforementioned techniques in different vascular beds is predictive of clinical events [39–54]. The principal studies are shown in Table 13.2. Some of these studies included type 2 diabetic patients. In type 2 diabetic patients with normal coronary arteries, coronary artery dysfunction, as assessed by cold-pressor test, was found to predict long-term cardiovascular outcomes, with a relative risk of 4.9 [49]. In a study of asymptomatic type 2 diabetic patients who underwent myocardial perfusion imaging, normal endothelial function had a 93 % negative predictive value in excluding CAD [50]. In patients with newly diagnosed CAD and impaired brachial artery FMD (40 % with diabetes), a persistently impaired FMD was an independent predictor of future cardiovascular events after six months of optimized lifestyle changes and pharmacotherapy [52]. A community based study in 1,016 older adults (72 % with diabetes) demonstrated that impaired forearm endothelial-dependent vasodilation was associated with a five year risk of major adverse cardiovascular events [54]. A recent meta-analysis of 14 observational studies and a recent review, both concluded that FMD is predictive of cardiovascular events and provides prognostic information that is at least equal to the information gained from conventional cardiovascular risk factors; however, future research is required to confirm FMD’s efficacy in the assessment of CVD risk [55, 56].
Table 13.2
Selected studies evaluating endothelial dysfunction as a predictor of cardiovascular events
Study (year) | n | Patient population | Arterial bed | Technique(s) | Endpoints | Mean follow-up (months) | Endothelial dysfunction as an independent predictor | Estimated RR of CV events |
|---|---|---|---|---|---|---|---|---|
Papaioannou et al. [50] | 75 | T2DM patients without CAD | Brachial | FMD, NMD, MPI | CAD | 60 | No association | na |
Nitenberg et al. [49] | 124 | Patients with HT or T2DM and normal coronary arteries | Coronary | CPT | CAD,CVD | 112 | Yes | 4.9 |
Nitenberg et al. [48] | 72 | T2DM patients without CAD | Coronary | CPT | CAD,CVD | 45 | Yes | 2.8 |
Chan et al. [46] | 152 | Patients with CAD | Brachial | FMD | CAD, CVD | 34 | Yes | 4.7a |
Carotid | IMT | |||||||
Halcox et al. [44] | 308 | Patients with and without CAD | Coronary | Acetylcholine response | CAD,CVD | 46 | Yes | 1.4 |
Perticone et al. [43] | 225 | Patients with untreated hypertension | Forearm | FABF | CAD,CVD, PVD | 32 | Yes | 2.1 |
Schachinger et al. [41] | 147 | Patients with chest pain or SVD | Coronary | Acetylcholine response, CPT, FMD, NMD | CAD,CVD, PVD | 80 | Yes | na |
Al Suwaidi et al. [39] | 157 | Patients with mild CAD | Coronary | Acetylcholine, adenosine, and nitroglycerin responses | CAD | 28 | na | na |
Muiesan et al. [51] | 172 | Patients with uncomplicated hypertension (28 % with diabetes) | Brachial | FMD | CV events | 109 | Yes | 2.5b |
Yeboah et al. [53] | 3,026 | Population based cohort of without known CVD | Brachial | FMD | CV event | 60 | Yes | na |
Kitta et al. [52] | 251 | Patients with CAD and optimized therapy (40 % with diabetes) | Brachial | FMD | CVD | 36 | Yes | 1.8c |
Lind et al. [54] | 1,016 | Community based study of older adults (>70 years), 72 % with diabetes | Forearm, Brachial, Radial, Carotid | Acetylcholine and sodium nitroprusside, FMD, PWA, IMT | CV disease (MI or stroke | 60 | Yes, Forearm EDV only | na |
Pathogenesis of Endothelial Dysfunction in Type 2 Diabetes Mellitus
Endothelial dysfunction has been demonstrated in T2DM in both the resistance and conduit vessels of the peripheral circulation [57–61], as well as in the coronary circulation [62, 63]. Plasma levels of the soluble adhesion molecules E-selectin, vascular cellular adhesion molecule (VCAM)-1 and intercellular adhesion molecule (ICAM)-1 are elevated in subjects with T2DM [3, 29, 30, 64]. Similarly, increased plasma levels of von Willebrand factor (vWF), a measure of endothelial cell damage and activation, are found in diabetes [3, 30, 64]. Microalbuminuria is an independent predictor of ED and may indicate widespread vascular dysfunction in diabetes [3, 65].
The precise pathogenetic mechanisms underlying the development of ED in T2DM remain unclear, but at inception they probably involve uncoupling of both eNOS activity (Fig. 13.1b), and mitochondrial oxidative phosphorylation (Fig. 13.2), as well as the activation of vascular NAD(P)H oxidase. These three mechanisms essentially result in increased generation of superoxide (O2 •−) radicals, eNOS uncoupling, and the overproduction of peroxynitrite. The main factors that combine to cause these biochemical disturbances are dyslipoproteinemia, oxidative stress [4], and inflammation [5–7]. Additional clinical factors that may contribute, either individually or synergistically, to ED in T2DM include hypertension [66], visceral obesity [67], insulin resistance [5, 68, 69], postprandial hyperlipidemia [70–72], fasting and postprandial hyperglycemia [73–75] and elevated levels of ADMA [37, 76].
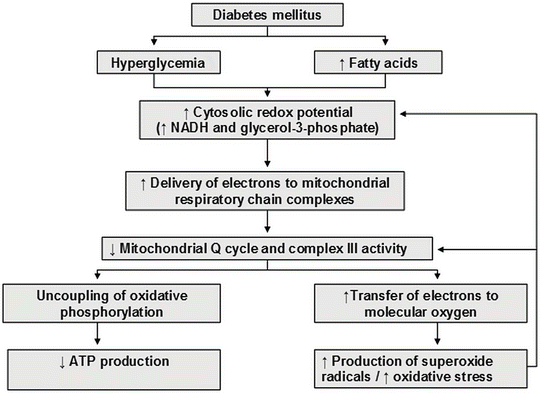
Fig. 13.2
Mechanism whereby hyperglycemia and elevated fatty acids induce uncoupling of mitochondrial oxidative phosphorylation and increased oxidative stress in diabetes. ATP adenosine triphosphate, NADH reduced nicotinamide adenine dinucleotide
The impact of insulin resistance in T2DM operates at an insulin signalling level in endothelial cells and in adipose tissue and skeletal muscle [5]. Impaired insulin receptor substrate-1 (IRS-1) and phosphatidylinositol (PI) 3-kinase insulin signalling results in decreased production of NO and ED on the one hand, and decreased glucose transporter (GLUT4) translocation and peripheral insulin resistance on the other. Insulin resistance also increases fatty acid availability which uncouples mitochondrial function in endothelial cells. This generates ROS by increasing advanced glycation end-products (AGES), protein kinase C (PKC) and N-acetylglucosamine (glcNAC), impairing eNOS activity and inducing ED. Inflammation, lipotoxicity and glucotoxicity are all increased in diabetes and collectively contribute to insulin resistance and ED [5]. Figure 13.3 suggests that the pathogenesis of ED in T2DM has oxidative stress as the central pathway for a wide spectrum of risk factors [3].
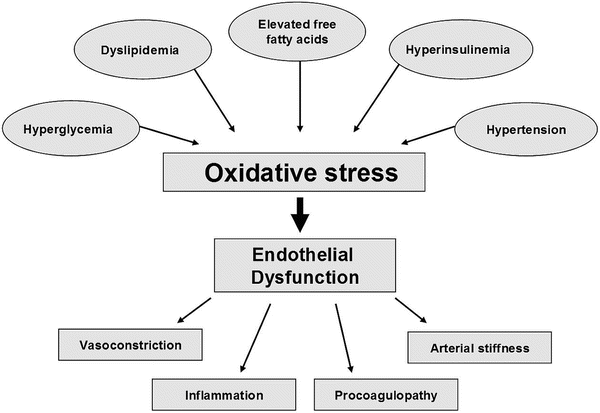
Fig. 13.3
Pathogenesis and consequences of endothelial dysfunction in type 2 diabetes mellitus. Oxidative stress also contributes to endothelial dysfunction by activating protein kinase C, polyol, hexosamine and NFkappa B pathways, as well as by increasing asymmetric dimethylarginine and advanced glycation end-products
Treating Endothelial Dysfunction in Type 2 Diabetes
Strategies for treating ED in T2DM will necessarily target the pathophysiological factors that underlie endotheliopathy, such as hyperglycemia, insulin resistance, dyslipidemia, increased oxidative stress, inflammation and hypertension [77, 78]. Treatment options range from lifestyle interventions to nutritional supplements and specific pharmacological therapies. The results of selected intervention studies are summarized in Table 13.3.
Table 13.3
Randomized controlled trials investigating the therapeutic regulation of endothelial function in patients with type 2 diabetes mellitus
Study (year) | n | Treatment | Treatment duration (months) | Endpoint | Treatment effect |
|---|---|---|---|---|---|
Lifestyle interventions | |||||
Maiorana et al. [80] | 16 | Aerobic and resistance exercise | 2 | FMD, ACh stimulated FABF | + |
Antiglycaemic agents and insulin sensitizers | |||||
Bagg et al. [91] | 43 | Improved glycemic control/usual gylcaemic control | 5 | FMD | ns |
Rask-Madsen et al. [97] | 28 | Insulin therapy/no hypoglycaemic drug theapy | 2 | Insulin stimulated FABF response to Ach | + |
Sundaresan et al. [98] | 14 | Metformin/glibenclamide | 1 | FABF response to diazoxide, ACh, SNP, norepinephrine | ns |
Williams et al. [99] | 14 | Glibenclamide/placebo | 1 | FABF response to Ach | ns |
Spallarossa et al. [100] | 20 | Glibenclamide/glimepriride/diet | 2 | FMD | ns |
Wascher et al. [101] | 15 | Gliclazide/glibenclamide | 1 | FABF response to hyperaemia | |
Abbink et al. [102] | 24 | Glibenclamide/glimepiride or metformin | 2 | FABF response to diazoxide, ACh, dipyridamole, forearm ischaemia | ns |
Manzella et al. [103] | 16 | Rapaglinide/glibenclamide | 2 | FMD | +a |
Schmoelzer et al. [104] | 12 | Rapaglinide/control | 2 h | FMD | + |
Mather et al. [105] | 43 | Metfromin/placebo | 3 | FABF response to Ach | + |
Natali et al. [106] | 74 | Rosiglitazone, Metformin, or placebo | 4 | FABF response to Ach | +b |
Caballero et al. [108] | 87 | Troglitazone/placebo | 3 | FMD | +c |
Martens et a l [110] | 20 | Pioglitazone/Placebo | 1 | FMD | + |
Pistrosch et al. [111] | 12 | Rosiglitazone/nateglinide | 3 | FABF response to Ach | +b |
Shimabukuro et al. [112] | 14 | Acarbose/placebo prior to a test meal | 1 week | FABF response to hyperemia | + |
Nystrom et al. [114] | 12 | Recombinant glucagon-like peptide-1/saline | 1 week | FMD | + |
Koska et al. [113] | 28 | Exenatide/placebo | Single dose | PAT | + |
Antihypertensive agents | |||||
O’Driscoll et al. [126] | 10 | Enalapril/placebo | 1 | FABF response to Ach | + |
Cheetham et al. [128] | 9 | Losartan/placebo | 1 | FABF response to Ach | + |
Cheetham et al. [129] | 12 | Losatan/placebo | 1 | FMD | + |
Ceriello et al. [127] | 20 | Atorvastatin/irbesartan/placebo | 1 week | FMD | + |
Flammer et al. [130] | 13 | Losartan/atenolol | 1 | FMD | +d |
Davies et al. [131] | 42 | Spironolactone/placebo | 1 | FABF response to Ach | − |
Yilmaz et al. [136] | 108 | Valsartin/amlodipine/valsartin + amlodipine | 3 | FMD | +e |
Antioxidants and nutritional supplements | |||||
Watts et al. [139] | 40 | CoQ10/placebo | 3 | FMD | + |
Playford et al. [140] | 20 | CoQ10/placebo | 3 | FABF response to ACh, BK, SNP | ns |
Lim et al. [141] | 80 | CoQ10/placebo | 3 | Microcirculatory function | ns |
Anderson et al. [144] | 20 | Vitamin C/placebo | 2 week | FMD | + |
Paolisso et al. [146] | 40 | Vitamin E/placebo | 2 | FMD | + |
Darko et al. [150] | 35 | Vitamin C/placebo | 3 week | FABF response to Ach | ns |
Tousoulis et al. [149] | 41 | Vitamin C/atorvastatin/placebo | 4 week | FABF response to hyperaemia | +f |
Chen et al. [151] | 32 | Vitamin C/placebo | 1 | FABF response to ACh, SNP or insulin | ns |
Gazis et al. [152] | 48 | α-tocopherol/placebo | 2 | FABF response to ACh, BK, SNP | ns |
Beckman et al. [153] | 23 | Vitamin E and C/placebo | 6 | FMD | ns |
Mangoni et al. [160] | 26 | Folic acid/placebo | 1 | FABF response to Ach | + |
Title et al. [161] | 19 | Folic acid/placebo | 2 week | FMD | + |
Miscellaneous therapies | |||||
Desouza et al. [162] | 14 | Sildenafil/placebo | 2 week | FMD | + |
Koh et al. [167] | 20 | Conjugated equine oestrogen/placebo | 2 | FMD | ns |
Silvestri et al. [168] | 30 | HRT/Tibolone/DHEAS | 1 | FMD | +g |
Howes et al. [169] | 16 | Red clover derived isoflavones/placebo | 1 | FABF response to Ach | ns |
Kwang et al. [177] | 35 | Testosterone/Placebo | 3 | FMD | + |
Bilsborough et al. [178] | 13 | Pentoxifylline/placebo | 2 | FABF response to ACh, FMD | ns |
Butler et al. [179] | 11 | Allopurinol | 1 | FABF response to Ach | + |
Lifestyle Interventions
Diet and exercise programs aimed at achieving weight loss improve many of the metabolic abnormalities in T2DM that contribute to ED, such as hyperglycemia, insulin resistance, visceral obesity, hypertension and dyslipidemia. Weight loss and increased physical activity have been shown to improve ED in type 2 diabetic patients. In an uncontrolled study, obese insulin resistant subjects (including subjects with T2DM), who underwent a six month lifestyle modification programme of caloric restriction and regular supervised exercise, achieved a 7 % mean reduction in body weight, with improvement in brachial artery FMD and reduction in markers of endothelial activation and coagulation [79]. Insulin sensitivity, glycemic control and HDL-cholesterol levels also improved.
A randomized, crossover study of combined aerobic and resistance exercise training for eight weeks demonstrated an increase in brachial artery FMD and acetylcholine (ACh)-stimulated forearm blood flow (FABF) in T2DM subjects [80]. Although glycemic control also improved, reductions in HbA1c and fasting glucose were not correlated with changes in endothelial function. Indeed, it appears that the benefits of exercise in improving ED are not necessarily dependent on improvement in traditional cardiovascular risk factors [81], suggesting that repeated exercise may also act directly on the vasculature via a shear stress-related mechanism, possibly involving endothelial nitric oxide synthase (eNOS) up-regulation or reduced nitric oxide (NO) degradation by free radicals [82].
Epidemiological studies provide a large body of evidence supporting the association between cigarette smoking and cardiovascular events [83]. Cigarette smoking is also associated with the premature development of macrovascular and microvascular complications in patients with T2DM [84]. Cigarette smoke increases inflammation, thrombosis and oxidation of LDL-cholesterol, with experimental and clinical evidence supporting the notion that increased oxidative stress results in vascular dysfunction [83]. Both active and passive cigarette smoking are associated with a dose-related impairment of endothelial function [85–87]. Brachial artery FMD was assessed in current and former healthy young adult smokers [85]. Former male smokers, but not former female smokers, had higher FMD than current smokers, suggesting that endothelial function may improve with smoking cessation [85]. A larger randomized, placebo controlled study investigated the effects of five smoking cessation pharmacotherapies on brachial artery FMD in 1,504 subjects [88]. Despite a greater weight gain, FMD significantly improved in subjects who quit and remained abstinent at one year, but did not change in those who continued to smoke [88]. Studies assessing the effects of smoking cessation in T2DM patients are warranted.
Lipid-Regulating Therapy
Lipid regulating therapies improve diabetic dyslipidaemia; however, the various agents work via differing mechanisms, targeting to a greater or lesser degree the various aspects of the dyslipoproteinaemia. These therapies, through both lipid lowering effects and direct effects on the vasculature may improve ED. The mechanisms and vascular benefits of statins, fibric acid derivatives, nicotinic Antiglycemic acid (niacin) and omega-3 fatty acids are reviewed in more detail in a subsequent section.
Antiglycaemic Agents and Insulin Sensitizers
Hyperglycemia contributes to ED by multiple mechanisms, many of which result in increased oxidative stress [89, 90]. The effect of short-term blood glucose control on endothelial function was examined in poorly controlled T2DM subjects, who were randomized to improved glycaemic control (multi-agent therapy, including insulin, to achieve and maintain glycaemic targets) or usual treatment for 20 weeks: no difference in brachial artery FMD was found between the treatment groups [9].
Insulin Therapy
Insulin treatment not only reduces glycemia, but may also directly increase endothelial NO production through 1-phosphotidylinositol 3-kinase signalling [92]. In an uncontrolled study in T2DM subjects on oral hypoglycemic therapy, switching to pre-meal insulin lispro at a dose to maintain equivalent glycemic control, improved fasting and postprandial brachial artery FMD, an effect that was further augmented by concomitant vitamin C therapy [93].
In uncontrolled studies in T2DM patients treated with oral hypoglycemic therapy, the addition of insulin treatment improved glycemia control and brachial artery FMD [94] or forearm vascular reactivity [95, 96]. A randomized, controlled trial in T2DM subjects with ischemic heart disease showed that insulin therapy reduced HbA1c levels and improved insulin-stimulated, but not unstimulated, FABF response to ACh [97].
Sulphonylureas and Insulin Secretagogues
Sulphonylureas reduce glycemia by binding to specific (SUR1) receptors, resulting in closure of pancreatic beta-cell potassium-dependent ATP channels and stimulation of endogenous insulin secretion. However, controlled, crossover studies of glibenclamide therapy in T2DM subjects did not show any change in acetylcholine-stimulated FABF response compared with metformin or placebo [98, 99], and treatment with either glibenclamide or glimepiride did not alter brachial artery FMD compared with diet treatment alone [100]. One double-blind, randomized, crossover trial in T2DM subjects suggested that gliclazide reduced FABF responses to hyperemia compared with glibenclamide, possibly due to differential binding of these agents to sulphonylurea receptors [101]. However, another study did not show any difference between these two agents on ACh-stimulated FABF [102]. In a randomized crossover study, treatment with repaglinide (a short-acting insulin secretagogue), but not glibenclamide, increased brachial artery FMD in diet-treated T2DM subjects; improvement in endothelial function was correlated with changes in postprandial glycemia [103]. In subjects with impaired glucose tolerance, ED following a glucose challenge was related to the level of hyperglycemia. Reduction in the glycaemic response following a single dose of repaglinide, ameliorated ED in a glucose dependent manner [104].
Metformin
Although its main anti-hyperglycemic action is to suppress hepatic gluconeogenesis, possibly by stimulation of AMP-activated kinase pathways, metformin may also increase insulin sensitivity in peripheral tissues. In a placebo-controlled trial, metformin treatment increased ACh-stimulated FABF and insulin sensitivity in diet-treated T2DM patients [105]. However, another randomized, double-blind, placebo-controlled trial in T2DM patients failed to show improvement in insulin sensitivity or ACh-stimulated FABF with metformin therapy, despite improved glycemic control [106].
Thiazolidinediones
Thiazolidinediones improve insulin sensitivity and reduce glycemia via PPAR-gamma receptor-mediated effects on adipocytes resulting in decreased hepatic glucose output and increased peripheral glucose uptake by skeletal muscle [107]. In addition, as PPAR-gamma receptors are also present in the endothelium, vascular smooth muscle cells and macrophages, these agents may also have direct anti-inflammatory and anti-atherogenic effects on the vasculature.
In a randomized, double-blind, placebo-controlled trial, troglitazone increased brachial artery FMD in recently diagnosed T2DM subjects without macrovascular disease, but not in subjects with more long-standing disease or macrovascular complications [108]. In a small uncontrolled trial, pioglitazone-treated T2DM subjects showed improvement in brachial artery FMD, with a significant association between changes in FMD and insulin sensitivity [109]. In a randomized, double-blind, placebo-controlled, crossover study in T2DM subjects, pioglitazone was also shown to increase brachial artery FMD, but improvement in endothelial function was not correlated with favourable changes in plasma insulin, free fatty acids, adiponectin or C-reactive protein (CRP) [110]. In double-blind, crossover trials, rosiglitazone was shown to increase ACh-stimulated FABF in T2DM patients [106, 111].
Alpha-Glucosidase Inhibitors
Administration of a single dose of Acarbose, an alpha-glucosidase inhibitor that targets postprandial hyperglycemia, has been shown to attenuate postprandial impairment of hyperemic FABF response in diet-treated T2DM patients [112].
Incretins
Glucagon-like peptide (GLP)-1 is an incretin that reduces glycemia by stimulating insulin secretion, suppressing glucagon secretion and slowing gastrointestinal motility. Gliptins inhibit dipeptidyl peptidase-4, thereby increasing incretin levels which in turn increases insulin secretion and decreases glycemia, predominantly through postprandial mechanisms [113]. In a randomized crossover study, infusion of recombinant GLP-1 was shown to increase brachial artery FMD in T2DM subjects, without any change in insulin resistance [114]. In a randomized crossover study, improved postprandial endothelial function following a single subcutaneous injection of exenatide (a DPP4 inhibitor) in T2DM patients was associated with decreased triglyceride but not glucose concentrations [113].
Amylin Agonists
Pramlintide, a synthetic amylin agonist, is associated with modest improvements in HbA1c levels and weight loss in insulin requiring T2DM patients. [115, 116] Pramlintide has also been shown to improve cardiovascular risk factors in T2DM patients: modest reductions in triglyceride levels [117] and improvement in markers of inflammation and oxidation have been reported [116, 117].
Other Emerging Therapies for T2DM
Type 2 sodium-glucose cotransporter inhibitors (SGLT2) have been shown to normalize glycemia by promoting renal glucose excretion in animal models [118]. Evidence suggests that succinobucol, a probucol analogue, has protective effects in diabetes via antiatherosclerotic, anti-inflammatory, antioxidant and potential antidiabetic activities [119].
Future longer-term cardiovascular outcome studies and postprandial arterial function studies, investigating the effects of incretins, gliptins, alpha-glucosidase inhibitors, amylin agonists, and succinobucol in statin-treated T2DM patients are warranted to establish if their effects translate to improved cardiovascular outcomes.
Antihypertensive Agents
In hypertension, increased oxidative stress and release of endothelial-derived constricting factors result in ED [8]. The coexistence of diabetes and hypertension has been shown to have an additive deleterious effect on endothelial function in the forearm resistance arteries [120]. Hyperglycemia increases the production of angiotensin II (Ang II) in the vessel wall [121]. Ang II stimulates vascular NAPH oxidase, increasing oxidative stress [122] and NF-kappaB activity, thereby activating inflammatory cytokines and vascular expression of cell adhesion molecules [9]. Hence, renin-angiotensin system (RAS) inhibition may improve endothelial function by reducing vascular oxidative stress and inflammation. Ang II may also promote release and production of vasoconstrictors such as endothelin-1 and prostaglandin-H2, which contribute to ED and hypertension.
Angiotensin-Converting Enzyme (ACE) Inhibitors
In a small uncontrolled study in hypertensive T2DM subjects, treatment with perindopril reduced blood pressure but did not improve methacholine-stimulated FABF [123]. However, a randomized, open parallel group study showed that quinapril treatment increased serotonin-stimulated FABF in T2DM subjects, perhaps by increasing vascular adiponectin expression [124]. In T2DM patients with proteinuria, improvement in brachial artery FMD following short term ramipril treatment was associated with a reduction in serum hsCRP and plasma long pentraxin 3 (PTX3) [125]. Furthermore, a double-blind, placebo-controlled crossover study showed that enalapril lowered blood pressure and improved ACh-stimulated FABF in T2DM subjects without vascular disease [126].
Angiotensin Receptor Antagonists
In randomized, controlled crossover trials, angiotensin type 1 receptor antagonists were shown to improve both FABF response to ACh and brachial artery FMD in subjects with T2DM [127–130]. Improvement in endothelial function occurred despite no significant change in blood pressure, and may relate to other treatment effects on oxidative stress, inflammation and endothelial cell activation.
Aldosterone Antagonists
On a cautious note, a randomized, double-blind, placebo-controlled trial showed that treatment with spironolactone worsened ACh-stimulated FABF in T2DM subjects, possibly due to worsening of glycemic control and increase in plasma Ang II [131].
Calcium Channel Blockers
Evidence for the effects of calcium channel blockade (CCB) on endothelial dysfunction is inconsistent. In a comparative study of antihypertensive agents, CCB (amlodipine) did not improve brachial artery FMD in patients with CAD [132]. In contrast, amlodipine improved endothelial function in hypertensive patients [133]. A study examining the effects CCB on endothelial function in hypertensive patients suggests a divergent effect for different types of these agents: efonidipine, a T- and L-type CCB, but not nifedipine, an L-type CCB, improved endothelial function and markers of oxidative stress [134]. Further, in patients with stable angina pectoris, combination CCB and ACE inhibition improved endothelial function, arterial stiffness and urinary albumin excretion more effectively than CCB alone [135]. However, in hypertensive T2DM patients with proteinuria, treatment with amlodipine, valsartan (an angiotensin II receptor blocker) or a combination of both, improved brachial artery FMD and proteinuria. Improvement in endothelial function was associated with reductions in PTX3 and soluble TNF-like weak inducer of apoptosis (sTWEAK) [136].
Antioxidants and Nutritional Supplements
Supplementation with antioxidants and/or factors essential to NO production may potentially improve ED in T2DM by re-coupling eNOS and mitochondrial function, as well as decreasing vascular NAD(P)H oxidase activity.
Increased oxidative stress in T2DM may disrupt coenzyme Q10 (CoQ10) composition and levels, resulting in defective antioxidant defences and further exacerbating oxidative stress and increasing membrane fluidity [14, 137, 138]. In endothelial cells this may lead to uncoupling of eNOS and a reduction in the release and subsequent activity of NO. CoQ10 as a potent antioxidant may decrease oxidative stress by not only quenching reactive oxidant species, but also by “recoupling” mitochondrial oxidative phosphorylation, thereby reducing superoxide production [14]. CoQ10 supplementation improved brachial artery FMD in treatment naive diabetic patients with dyslipidemia, but there was no change in glyceryl-trinitrate mediated endothelial-independent response, forearm vascular reactivity or plasma F2-isoprostanes [139, 140]. However, CoQ10 supplementation did not improve microcirculatory endothelial function in type 2 diabetic patients, despite repleted plasma CoQ10 concentrations [141].
Vascular responses to several other antioxidants and nutritional supplements have been examined, with inconsistent results being reported. Vitamin C (ascorbic acid) and vitamin E (tocopherol) have well-described antioxidant properties. However, studies examining their effect on ED in T2DM patients have yielded mixed results, some demonstrating benefit [142–148], while others have failed to show an effect [149–153]. Alpha-lipoic acid, another compound with free radical-scavenging activity, was shown to improve ACh-stimulated FABF [143]. Despite the potential for vascular benefit with the polyphenolic antioxidants present in red wine [154, 155], benefit has not been demonstrated in T2DM patients [156]. Supplementation with l-arginine, a principal substrate for eNOS, improved both brachial artery FMD and post-ischaemic forearm hyperaemia in T2DM women [148]. Oxidation of tetrahydrobiopterin (BH4) may lead to uncoupling of eNOS, reducing NO production and further generating oxidant species. Intra-arterial BH4 infusion was shown to improve FABF response to ACh in T2DM subjects [157]. Folic acid, a strong peroxynitrite scavenger, may also protect BH4 from oxidation, reversing eNOS uncoupling [158]. Folic acid has been shown to improve FABF and brachial artery FMD in T2DM patients [159–161].
Miscellaneous Therapies
Phosphodiesterase Inhibitors
The vasorelaxation effect of NO on vascular smooth muscle is mediated by cyclic GMP (cGMP), which is catabolised by phosphodiesterase (PDE). PDE inhibitors, which are used to treat erectile dysfunction, increase the bioavailability of cGMP, which activates protein kinase G thereby promoting vasodilatation and a penile erection. Sildenafil, a selective PDE-5 inhibitor, has been shown to increase brachial artery FMD in a double-blind, placebo-controlled, crossover study in T2DM men with erectile dysfunction [162].
Estrogen Therapy
Epidemiological studies have suggested a protective effect of estrogen on cardiovascular risk, but intervention trials of sex hormone replacement in post-menopausal women have reported no benefit, and even a possible initial adverse effect, on cardiovascular outcomes [163, 164]. Although estrogen therapy may protect endothelial function by up-regulating endothelial NO production, reducing the formation of cyclooxygenase (COX) derived endothelium-derived contracting factors and have favorable effects on lipids and blood pressure, it may also have adverse effects in increasing vascular inflammation and cell adhesion [8, 165]. The effect of hormone replacement therapy on endothelial function in post-menopausal T2DM women has been inconsistent [166–169].
Testosterone Therapy
In men with testosterone deficiency brachial artery FMD has been reported to be both increased [170, 171] and impaired [172–174]. Testosterone deficiency is associated with elevated triglyceride and low HDL-cholesterol concentrations [175] and this could explain its association with impaired endothelial function. Evidence for the effect of testosterone replacement on endothelial function is inconsistent, however. Testosterone replacement for 12 weeks reduced FMD in hypogonadal men [171]. In a small study, hypogonadal men were found to develop impaired FMD four weeks following testosterone pellet implantation [176]. In contrast, in a randomized, placebo-controlled study, 12 weeks of testosterone replacement improved brachial artery reactivity in men with CAD [177]. Studies of the effect of testosterone replacement on endothelial function in diabetic men with androgen deficiency are warranted.
Tumour Necrosis Factor (TNF)-Alpha Inhibitors
The pro-inflammatory cytokine tumour necrosis factor (TNF)-alpha may contribute to ED by stimulating vascular NADPH oxidase and increasing superoxide production and oxidative stress. However, in a randomized crossover study, pentoxifylline, an inhibitor of TNF-alpha production, did not alter ACh-stimulated FABF in T2DM subjects, despite reduction in serum TNF-alpha levels [178].
Xanthine Oxidase Inhibitors
Xanthine oxidase is an enzyme present in endothelial cells that when activated increases oxidative stress. In a small randomized placebo-controlled trial allopurinol, an inhibitor of xanthine oxidase, was shown to improve ACh-stimulated FABF, and hence resistance artery function in mildly hypertensive T2DM subjects. There was no reduction in blood pressure, however [179].
Diabetic Dyslipidemia
Pathogenesis of Diabetic Dyslipidemia in Type 2 Diabetes
Dyslipidemia is a common risk factor and a strong predictor of CVD in T2DM patients [180]. Elevated plasma concentrations of triglycerides and reduced high-density lipoprotein (HDL)-cholesterol, in both the fasting and postprandial states, are the major lipoprotein abnormalities in diabetic dyslipidemia. The accumulation of small dense low-density lipoprotein (sdLDL) particles and triglyceride-rich lipoproteins (TRLs), including chylomicron remnants and very-low density lipoprotein (VLDL) remnants, are also characteristic of the atherogenic lipid profile [181–184]. These abnormalities are reflected by increased plasma concentrations of non-HDL cholesterol and apolipoprotein B-100 (apoB) [182]. In the postprandial state there is an increase in plasma TRLs and their remnants and qualitative changes in low-density lipoprotein (LDL) and HDL particles [181]. Therefore, hypertriglyceridemia is a marker of a range of lipoprotein abnormalities not routinely measured in clinical practice [182].
The aetiology of diabetic dyslipidemia is complex [185, 186]. It relates collectively to hyperglycemia [187], insulin resistance [187], hyperinsulinemia [187, 188], abdominal visceral adipose disposition and increased liver fat content [188], and dysregulated fatty acid metabolism [188]. Insulin resistance increases fatty acid flux from visceral adipose tissue to the liver, inducing increased liver fat content, over production of VLDL1 particles [181, 188] and a reduction in the inhibitory effect of insulin on hepatic apoB secretion [189–191]. Hyperglycemia further aggravates the overproduction of VLDL1, in particular increased VLDL1 triglyceride production rate [187]. Collectively, plasma glucose, insulin and free fatty acids explain approximately half of the variation in VLDL1 production rate [187].
Impaired chylomicron clearance in T2DM results from the reduced activity of lipoprotein lipase (LPL), an endothelial bound enzyme, and decreased receptor-mediated endocytosis in the liver [181, 192, 193]. VLDL1 particles compete with chylomicrons and its remnants for clearance by saturating the lipolytic capacity of LPL and the activity of hepatic receptors [181, 192]. Hepatic secretion of apolipoprotein CIII (apoCIII) is also increased in insulin resistance. This small protein, which is attached to VLDL, contributes to the delayed clearance of TRLs by inhibiting LPL and the binding of remnant TRLs to hepatic clearance receptors [192]. These mechanisms collectively account for postprandial lipaemia [181, 192] and may be an important causal mechanism of ED in T2DM and treatment targets for reducing residual cardiovascular risk.
Important compositional and atherogenic changes in lipoproteins are seen in T2DM [181]. An increased VLDL triglyceride pool leads to cholesterol depletion and triglyceride enrichment of LDL and HDL, mediated via the action of cholesteryl ester transfer protein (CETP) [181, 182]. Increased phospholipid transfer protein (PLTP) activity may contribute to hypertriglyceridemia and compositional changes in HDL. Further, the over activity of hepatic lipase, commonly increased in T2DM, increases the lipolysis of triglyceride enriched LDL and HDL particles [3, 4]. Compositional changes in HDL are also mediated by the actions of lipoprotein lipase (LPL) [181]. Collectively, these compositional changes produce smaller and denser lipoprotein particles that are potentially more atherogenic. Small dense LDL particles more easily penetrate the arterial wall and have a higher binding affinity to intimal proteoglycans than more buoyant larger LDL particles [3, 181, 194, 195]. In the intima, retained LDL particles are modified when exposed to oxidative stress, with sdLDL having an increased sensitivity to oxidation; glycation of LDL further increases this susceptibility to oxidation [181]. A predominant feature of diabetic dyslipidemia is low HDL-cholesterol concentrations with greater reductions in HDL2 than HDL3 [181]. In parallel to these reductions in HDL particles are reductions in plasma levels of apolipoprotein A-I (apoA-I) and apoA-II and HDL lipoproteins containing both Apo A-I and Apo A-II (LpA-I:A-II) [3, 181, 196]. These compositional changes in HDL particles are important in respect to ED and atherogenicity, as they are associated with a reduction in rates of reverse cholesterol transport and a decrease in the direct anti-atherogenic effects of HDL, including its anti-oxidant, anti-inflammatory and anti-thrombotic effects [3, 197–199]. These lipoprotein abnormalities and the associated risk of ED and cardiovascular disease in T2DM may be determined by various genes that regulate lipid and lipoprotein functionality, for example, LPL [200], apolipoprotein E [201], apoC-III [202, 203] and CETP [204]. This suggests a connection for the genetic control of ED in T2DM [3].
Treatment of Diabetic Dyslipidemia
Current clinical guidelines emphasize lifestyle modifications and pharmacotherapy for the reduction of dyslipidemia and CVD risk in T2DM patients [205, 206].
Lifestyle Interventions
Initial management should include an individualized lifestyle modification programme to optimize weight loss and glcyemic control. Benefits of weight reduction in T2DM increase steadily with increasing weight loss and include reductions in waist circumference, blood pressure, fasting glucose, HbA1c and serum triglycerides, resulting in improved metabolic control and CVD risk factor reduction [207, 208]. In the Look AHEAD (Action for Health in Diabetes) study, weight loss and improved physical fitness in type 2 diabetic patients was associated with improved glcyemic control and CVD risk factor reduction [208, 209]. In type 2 diabetic patients, a reduction in insulin resistance and fat mass following prolonged aerobic exercise resulted in improvements in lipoprotein metabolism [210].
Lipid Regulating Therapy
Should dyslipidemia persist following a trial of intensified lifestyle changes, the next approach is pharmacotherapy, either an intensification of statin therapy or the addition of a second lipid regulating agent.
Hydroxymethylglutaryl (HMG)-CoA Reductase Inhibitors (Statins)
Patients with an increasing number of metabolic syndrome components, with or without diabetes, have a progressive risk of CVD, and derive greater incremental benefit from higher dose statin therapy [211]. Statin therapy in hypertriglyceridemic patients, with and without T2DM, reduces triglyceride concentrations by up to 45 % [212–216], in a dose dependent manner and proportional to LDL-cholesterol lowering [215, 216]. Statin-treated patients with combined low LDL-cholesterol (<1.8 mmol/L) and low triglyceride (<1.7 mmol/L) levels had the lowest CHD event rate in the PROVE IT-TIMI 22 trial [217]. Evidence suggests that statins may mediate triglyceride lowering in T2DM by increasing the catabolism of TRL- triglyceride [213] and the TRL’s VLDL1-ApoB, VLDL2-ApoB and IDL-ApoB [218]. Statins may further mediate triglyceride lowering by reducing the production rate and secretion of VLDL1-ApoB [218].
Fibric Acid Derivatives
A recent meta-analysis concluded that fibrates are effective in reducing CVD events, primarily by prevention of coronary events [219]. The lipid-regulating effects of fibrates, mediated via PPAR-alpha receptor, are predominantly to promote fatty acid catabolism and reverse cholesterol transport, resulting in triglyceride lowering and increase in HDL-cholesterol and LDL particle size [107]. In a subgroup analysis of the Helsinki Heart Study, diabetic patients when compared with non-diabetic subjects were more dyslipidemic, at higher CVD risk and achieved a modest but non-significant reduction in CVD risk with gemfibrozil therapy [220]. The Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study demonstrated that fenofibrate reduced CVD events in hypertriglyceridemic T2DM patients with or without low HDL-cholesterol [221]. Indeed, in FIELD a post hoc analysis showed that the greatest benefit of fenofibrate therapy was seen in patients with triglycerides ≥2.3 mmol/L and reduced HDL-cholesterol; a relative risk reduction of 27 % was demonstrated [221].
The benefits of combined fibrate and statin therapy was examined in the Action to Control Cardiovascular Risk in Diabetes (ACCORD)-Lipid study. In ACCORD-Lipid, 5,518 simvastatin-treated patients with diabetes were randomized to receive either fenofibrate or placebo. Compared to simvastatin alone, 4.7 years of combination therapy did not reduce the rate of major fatal or nonfatal cardiovascular events [222]. However, in a valid pre-specified analysis of patients with triglycerides >2.3 mmol/L and HDL-cholesterol <0.9 mmol/L (approximately 17 % of the ACCORD-Lipid population), combined therapy achieved an additional 31 % reduction in cardiovascular risk, though this did not achieve statistical significance [222]. ACCORD Lipid did not support the use of combined statin and fenofibrate therapy in the majority of T2DM patients [222], but in those who have hypertriglyceridemia with or without low HDL-cholesterol, despite intensification of statin therapy, adding a fibrate may be beneficial in reducing residual CVD risk.
Nicotinic Acid (Niacin)
Niacin is one of the oldest lipid regulating therapies and remains the most potent therapy available for increasing HDL-cholesterol [223, 224]. Many of niacin’s effects are thought to derive from its action on adipose tissue [225]. However, the cellular mechanism for Niacin’s lipid-lowering effects were not fully elucidated until the identification in 2003 of a G protein-coupled receptor GPR109A (HM74A), which is highly expressed in adipose tissue and acts as a high affinity receptor for nicotinic acid and mediates it antilipolytic effects [223, 226–229]. By binding to GPR109A, niacin inhibits hormone-sensitive lipase activity, resulting in decreased free fatty acid (FFA) release from adipose tissues. This results in a decreased flux of FFA to the liver that may reduce triglyceride production and subsequent hepatic VLDL production [226, 230, 231]. Niacin may also directly and non-competitively inhibit hepatic diacylglycerol acyl transferase (DGAT-2), the key enzyme in triglyceride synthesis [231, 232]. Although niacin increases HDL-cholesterol by up to 30 % (at therapeutic doses), a mechanism for this HDL raising effect remains to be fully elucidated but it may involve the down-regulation of the HDL-catabolism receptor [224, 230, 233, 234]. The lipid-regulating effects of niacin may be further mediated through PPAR-mediated transcriptional regulation and may involve all three PPAR isoforms, alpha (α), gamma (γ) and delta (δ) [225].
Niacin has been shown to be effective in lowering cardiovascular risk when used as monotherapy [182, 235–237], with recent post hoc analyses of the Coronary Drug Project demonstrating that the benefits were independent of hyperglycemia, metabolic syndrome and diabetes [182, 237–239]. Combination therapy with a stain and niacin is associated with regression of coronary atherosclerosis and carotid intima–media thickness (CIMT) in patients at high cardiovascular risk with low HDL-cholesterol levels, including those with diabetes [240–243]. These beneficial effects may reflect the reduction in triglycerides and increase in HDL-cholesterol seen with ER Niacin [182].
However, the question of whether adding niacin to statin therapy translates to improved cardiovascular outcomes in T2DM remains unanswered. The Atherothrombosis Intervention in Metabolic Syndrome with Low HDL-cholesterol/High Triglyceride and Impact on Global Health Outcomes (AIM-HIGH) study was discontinued after 36 months of follow-up due to lack of clinical benefit from the addition of ER Niacin to statin (± ezetimibe) therapy, despite significant improvements in HDL-cholesterol and triglycerides [244]. The Heart Protection Study-2 and the Treatment of HDL to reduce the incidence of vascular events (HPS2-THRIVE), due to report in 2013, will address if adding ER Niacin to statin therapy reduces CVD events in high-risk patients with prior vascular disease, a significant proportion having the metabolic syndrome or diabetes [245].
Omega-3 Fatty Acids
Supplementation with omega-3 fatty acid ethyl esters (eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)), at doses of 3–4 g daily, lower plasma triglycerides particularly in patients with hypertriglyceridemia [182, 192]. Evidence suggests combined statin and omega-3 fatty acid therapy may reduce cardiovascular events. In the Gruppo Italian per lo Studio della Sopravvi-venza nell’Infarto Miocardio GISSI-Prevenzione (GISSI-Prevenzione) study, combination EPA/DHA dosed at 1 g daily reduced all cause mortality and sudden death in subjects with previous myocardial infarction [246]. The Japan Eicosapentaenoic Acid Lipid Intervention Study (JELIS) demonstrated a reduction in major cardiovascular events with combined omega-3 fatty acids (EPA 1,800 mg daily) and low dose statin (pravastatin 10 mg or simvastatin 5 mg daily) compared to statin alone [247]. However, this cardiovascular benefit may relate in part to the antiarrhythmic effects of omega-3 fatty acids and is independent of minor changes in plasma triglycerides [182, 192]. A comprehensive review of the cardiovascular effects of omega-3 fatty acids has recently been published [248]. Importantly, there is no published outcome evidence demonstrating the beneficial effects of treating residual hypertriglyceridemia with omega-3 fatty acid therapy in statin-treated T2DM patients [182].
Cholesteryl Ester Transfer Protein (CETP) Inhibition
Cholesteryl ester transfer protein (CETP) inhibitors have the potential to correct diabetic dyslipidemia by inhibiting the heteroexchange of neutral lipids among lipoproteins in both fasting and postprandial states. In subjects with low HDL-cholesterol concentrations, 4 weeks of torcetrapib treatment, alone or in combination with atorvastatin, resulted in a dose-related increase in plasma HDL-cholesterol and reductions in triglyceride and LDL-cholesterol concentrations [249, 250]. The off-target effects of torcetrapib on blood pressure are not seen with other CETP inhibitors, such as anacetrapib, dalcetrapib and evacetrapib, so that this class of agents may still have a role as second line agents in the management of diabetic dyslipidemia [251]. However, the preliminary results from the Dal-VESSEL study were reported as showing no effect of dalcetrapib on FMD, but the Dal-PLAQUE study [252] suggested a small trend to improvement in plaque volume; diabetic subjects were not specifically studied and the effect on clinical endpoints must await publication of the Dal-OUTCOMES study. The effect of CETP inhibitors on endothelial function in diabetes warrants investigation, however.
Evidence That Lipid Regulation Improves Endothelial Function in Diabetes
The rationale for lipid regulation of diabetic dyslipidemia is well supported by studies showing correction of endothelial dysfunction. The results of selected intervention studies utilizing lipid regulating therapy are reviewed below and summarized in Table 13.4.
Table 13.4
Randomized controlled trials investigating the therapeutic regulation of endothelial function in patients with type 2 diabetes mellitus: Lipid regulating therapies
Study (year) | n | Treatment | Treatment duration (months) | Endpoint | Treatment effect |
|---|---|---|---|---|---|
Lipid regulating agents | |||||
Statins | |||||
Tsunekawa et al. [260] | 27 | Cerivastatin/Placebo | 3 days | FMD | + |
Tan et al. [261] | 80 | Atorvastatin/Placebo | 6 | FMD | + |
Ceriello et al. [262] | 30 | Simvastatin/Placebo | 3–6 days + 3 | FMD | + |
Ceriello et al. [127] | 20 | Atorvastatin/irbesartan/placebo | 1 week | FMD | + |
Dalla et al. [263] | 25 | Atorvastatin/placebo | 12 | VCAMI and E-selectin | + |
Brunetti et al. [265] | 22 | Atorvastatin/rosuvastatin | 3 | FMD | + |
Adel et al. [266] | 60 | Atorvastatin/placebo | 4 week | FMD | + |
Economides et al. [267] | 40 | Atorvastatin/placebo | 3 | FMD | ns |
Beishuizen et al. [268] | 250 | Cerivastatin replaced by simvastatin | 24 | FMD | ns |
Van Venrooij et al. [269] | 133 | Atorvastatin 10 mg/Atorvastatin 80 mg | 7.5 | FMD | ns |
Tantikosoom et al. [271] | 42 | Atorvastatin/placebo | 7.5 | FMD | ns |
Tousoulis et al. [149] | 41 | Atorvastatin/Vitamin C/No treatment | 1 | FABF response to post-ischaemic hyperemia | +a |
Tran et al. [272] | 11 | Cerivastatin/placebo | 2 | FABF response to ACh and L-NMMA | ns |
Fibrates | |||||
Playford et al. [281] | 40 | Fenofibrate/placebo | 3 | FMD | + |
Playford et al. [140] | 20 | Fenofibrate/placebo | 3 | FABF response to ACh, BK, SNP | ns |
Evans et al. [283] | 20 | Ciprofibrate/placebo | 3 | FMD | + |
Avogaro et al. [284] | 10 | Gemfibrozil/placebo | 3 | FMD | + |
Fegan et al. [285] | 10 | Fenofibrate/placebo | 3 | Blood flow responses to iontophoresis of ACh | ns |
Hiukka et al. [286] | 170 | Fenofibrate/placebo | 60 | IMT, AIx, biomarkers of endothelial activation | ns |
Niacin | |||||
No studies identified in T2DM patients | |||||
Omega-3 fatty acids | |||||
McVeigh et al. [294] | 23 | Fish oil/placebo | 1.5 | FABF response to ACh | + |
Woodman et al. [295] | 51 | EPA/DHA/placebo | 1.5 | FMD | ns |
West et al. [296] | 18 | MUFA ± omega-3 FA | Three test meals over 3 week | FMD | + |
Combination therapies | |||||
Hamilton et al. [310] | 15 | Statin + fenofibrate/statin + placebo | 3 | FMD + forearm microcirculatory function | + |
Fegan et al. [285] | 11 | Cerivastatin + fenofibrate | 3 | Skin blood flow response to iontophoresis and skin maximum hyperaemia | ns |
Lee et al. [315] | 71b | Statin + Niacin/statin/placebo | 12 | Carotid MRI, aortic distensibility, MRI brachial artery FMD | +c |
Hamilton et al. [316] | 15 | Statin + Niacin/statin alone | 5 | Small artery vasodilation and compliance | + |
Hamilton et al. [320] | 23 | Statin+ CoQ10/statin + placebo | 3 | FMD | + |
Koh et al. [322] | 50 | Simvastatin + ramipril | 2 | FMD | ++ |
Ceriello et al. [127] | 20 | Atorvastatin + irbesartan | 1 week | FMD | ++ |
Playford et al. [140] | 20 | Fenofibrate + CoQ10 | 3 | FABF response to ACh, BK, SNP | + |
Luescher et al. [331] | 476d | Dalcetrapib + statin/placebo + statin | 9 | FMD | ns |
Hydroxymethylglutaryl (HMG)-CoA Reductase Inhibitors (Statins)
Statins, inhibitors of hydroxymethylglutaryl (HMG)-CoA reductase, have been proven in large clinical trials to reduce cardiovascular mortality in a wide range of population subgroups, including subjects with diabetes [253]. Apart from their main effect in lowering LDL-cholesterol, statins may also have direct anti-inflammatory and antioxidant effects on the vasculature [77]. Statins have been shown to improve endothelial function in non-diabetic subjects with dyslipidaemia [254, 255], but results in T2DM subjects have been inconsistent and contradictory. Uncontrolled studies have not shown any benefit of statin therapy on serotonin-stimulated forearm blood flow (FABF) or brachial artery FMD in T2DM subjects [256–259]. One trial suggested improvement in endothelial function in a subgroup who achieved greater LDL-lowering [259], but another showed no benefit despite intensive lipid-lowering [257].
Related posts:
 Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
Insulin Resistance and Atherosclerosis
Niacin Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
The Role of Modified Forms of LDL and Corresponding Autoantibodies in the Development of Complications in Diabetes
Cholesterol Absorption Inhibitors (Ezetimibe) and Bile Acid Binding Resins (Colesevelam HCl) as Therapy for Dyslipidemia in Patients with Diabetes Mellitus
Roles of Extravasated and Modified Plasma Lipoproteins in Diabetic Retinopathy
Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
Insulin Resistance and Atherosclerosis
Niacin Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
The Role of Modified Forms of LDL and Corresponding Autoantibodies in the Development of Complications in Diabetes
Cholesterol Absorption Inhibitors (Ezetimibe) and Bile Acid Binding Resins (Colesevelam HCl) as Therapy for Dyslipidemia in Patients with Diabetes Mellitus
Roles of Extravasated and Modified Plasma Lipoproteins in Diabetic Retinopathy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


