Endocrine therapy for hormone receptor-positive breast cancer
Aman U. Buzdar, MD, FACP  Shaheenah Dawood, MD, MBBch, FACP, FRCP, MPH, CPH
Shaheenah Dawood, MD, MBBch, FACP, FRCP, MPH, CPH  Harold A. Harvey, MD
Harold A. Harvey, MD  Virgil Craig Jordan, OBE, PhD, DSc, FMedSci
Virgil Craig Jordan, OBE, PhD, DSc, FMedSci
Overview
The importance of the reproductive endocrine system in breast cancer treatment began to be appreciated at the turn of nineteenth century. It was around this time that it was realized that approximately one-third of premenopausal women with advanced breast cancer would respond to oophorectomy. However, it was only when the estrogen receptor (ER) was discovered that it was possible to fully appreciate the mechanisms underlying the activity of ovarian ablation and other associated treatments for breast cancer such as ovarian irradiation, adrenalectomy, and hypophysectomy. Research into both the estrogen and progesterone pathways not only provided a deeper understanding of the underlying mechanism of the carcinogenic pathway involved in the development of breast cancer but also allowed identification of potential targets for therapeutic intervention.
This chapter discusses recent advances in the molecular biology and physiology underlying the estrogen and progesterone receptor (PR) pathways and potential targets for intervention. In addition it examines and compares the pharmacology and efficacy of the different endocrine agents used in the management of both early and advanced stage breast cancer.
Biology of progestin production and action
Progestins are involved in the regulation of development and differentiation, proliferation, apoptosis, and metabolism in many target tissues with broad implications in neoplasia. In addition progestins serve as precursors to the estrogens, androgens, and adrenocortical steroids. Some of the progestin effects on target tissues are mediated by transcription, whereas other effects are more rapid and do not involve direct transcriptional effects. Progestins (Figure 1) include the naturally occurring hormone progesterone, 17α-acetoxyprogesterone derivatives in the pregnane series, 19-nortestosterone derivatives (estranes), and norgestrel and related compounds in the gonane series. In humans progesterone is the most important progestin.
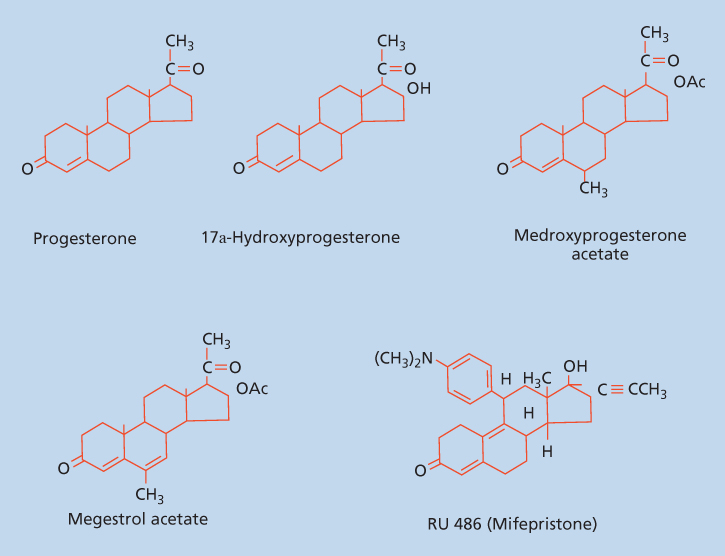
Figure 1 Structure of progesterone, 17α-hydroxyprogesterone, and synthetic progestins.
Synthesis and sites of production
Progesterone is produced early in the scheme of the synthetic pathway involving the conversion of cholesterol to androgens, progestins, and estrogens. After menopause, in the absence of hormone replacement, the adrenal gland becomes the principal source of progestins (through the conversion of pregnenolone) as well as other sex steroids. In the premenopausal woman, progesterone is principally derived from the corpus luteum of the ovary, but in pregnancy after the eighth week of gestation, placental progesterone production greatly exceeds ovarian-derived progesterone. The placental trophoblast is the dominant cell responsible for progesterone production by the placenta. The development of a secretory endometrium in which the blastocyst can implant requires progesterone. Progesterone levels of 25 ng/mL are usual in the luteal phase of the menstrual cycle, whereas levels up to 150 ng/mL are seen in late pregnancy.
Mechanism of action
Progesterone functions in RNA transcription regulation through a complex series of interactions that is initiated by binding of the hormone to its cognate receptor. There are two isoforms of the progesterone receptor (PR) known as PR-A and PR-B that have distinct biological activities. PR-B has been shown to mediate the stimulatory activities of progesterone, while PR-A functions to inhibit the action of PR-B as well as other steroid receptors.1–3 Both isoforms are encoded by a single gene, and their ratios vary in reproductive tissues as a consequence of developmental status, hormonal levels, and tissue type. Both isoforms of PR contain AF-1 and AF-2 transactivation domains with PR-B also containing an additional AF-3 domain which functions to contribute to its cell- and promoter-specific activity. The ligand for both isoforms of PR is identical.
In the absence of the hormone, PR is found in the nucleus in an inactive monomeric state associated with a complex of heat shock proteins (HSP -90, 70, 60, and 40) and is transcriptionally inactive.4, 5 Binding of progesterone to PR results in the dissociation of the heat shock proteins leading to the formation of receptor-ligand homodimers that remain localized in the nucleus and bind to highly selective progesterone response elements (PREs) located on target genes.6 It is important to note that target cells must distinguish not only progesterone from other steroids, present in small amounts, but also progesterone from other hydrophobic molecules that are frequently found in 100-fold or greater excess. Such a high degree of discrimination is limited to differentiated cells that possess PR proteins and activatable PREs in their genome.6, 7 The next step in the process is the transcriptional activation by PR, which results from the interaction with a number of coactivators including steroid receptor coactivator 1 (SRC-I), transcription intermediary factor 2, and retinoic acid coactivator 3, among others.6–9 The SRC-1 interacts with the N-terminal AF-1 and the C-terminal AF-2 of the PR. This serves to emphasize that SRC-1 function to synergize the ligand-independent amino terminal AF-1 with the ligand-responsive carboxyl terminal AF-2 of the PR. The PR-coactivator complex then interacts further with additional proteins that have histone acetylase activity that causes chromatin remodeling serving to increase accessibility of transcriptional proteins to the promoter target.8
Physiologic actions
Progestins are involved in a number of benign physiologic changes ranging from differentiated secretory activity to edematous changes in stromal tissues of the breast. They also have implications on neoplastic processes being associated with both a decreased risk of endometrial neoplasms and a slightly increased risk of breast neoplasms when used in conjunction with estrogen replacement for menopause. Progestins have a critical role in the support of the products of conception: the differentiation of the endometrium and the promotion of the secretory phase of the endometrium; the maturation and cornification of the vaginal mucosal epithelium; the suppression of ovulation; the inhibition of gonadotropin release; the proliferation of breast epithelium and the induction of secretory activity in breast epithelium; and a natriuretic effect on the kidneys. A number of these biologic effects of progesterone are seen only in concert with priming of the target tissues with estrogen, whereas other effects appear to be interrelated with the actions of other steroid hormones, peptide hormones, and/or growth factors. Possible interactions between progesterone, estrogen, and growth factors must be taken into account when constructing treatment strategies.
Synthetic progestins
Synthetic progestins are derivatives of the steroid structure of either progesterone or testosterone. The synthetic progestins most often encountered include 17-hydroxyprogesterone, medroxyprogesterone (Provera), medroxyprogesterone acetate (MPA), megestrol acetate (MA) (Megace), norethindrone, norethindrone enanthate, norethindrone acetate, norethynodrel, norgestrel, desogestrel, and gestodene. The two most widely used synthetic progestins are MPA and MA, which differ only by a single bond at C6–C7. MPA can be administered as either an oral or intramuscular (IM) preparation, while MA is administered as an oral preparation.
Synthetic progestins are used most frequently in contraception, in conjunction with estrogen in postmenopausal hormone replacement therapy (HRT) and in endocrine treatment of uterine and breast cancer. In postmenopausal HRT, progestins are added to estrogen replacement principally to minimize the risk of uterine cancer associated with estrogen-only therapy, although the effects of adding progestins to estrogen on the risk of breast cancer have become of increasing concern.10 In premenopausal women, progestins and antiprogestins are used predominantly in contraceptive preparations.11 Progestins are often used alone in selected women with climacteric symptoms who are advised not to take estrogens.
Metastatic breast cancers
Progestins and PR have been studied extensively in cancerous human breast tissue. Patients whose tumors are PR positive have a higher probability of responding to endocrine therapy (not necessarily progestins) and in most series show a somewhat better prognosis with respect to both survival and disease-free interval.12 Both MPA and MA have been shown to produce similar reductions in serum estrogen levels and produce responses of approximately 30% in patients with metastatic breast cancer.13 The principal progestin used for metastatic breast cancer has been MA. The response of metastatic breast cancer to MA is predicted not only by the presence of ER (estrogen receptor) and/or PR but also by the observation of an objective response to previous hormonal therapy. Randomized studies have shown comparable efficacy of progestins to tamoxifen, aminoglutethemide, and aromatase inhibitors in the second and subsequent lines of treatment of metastatic breast cancer.13, 14 Currently progestin therapy for hormone receptor-positive metastatic breast cancer is used principally after disease progression has been observed following use of selective ER modulators (e.g., tamoxifen) and aromatase inhibitors. As such a trial of progestins is commonly used as the third or subsequent line of therapy in patients with hormone receptor-positive metastatic breast cancer (Figure 2).
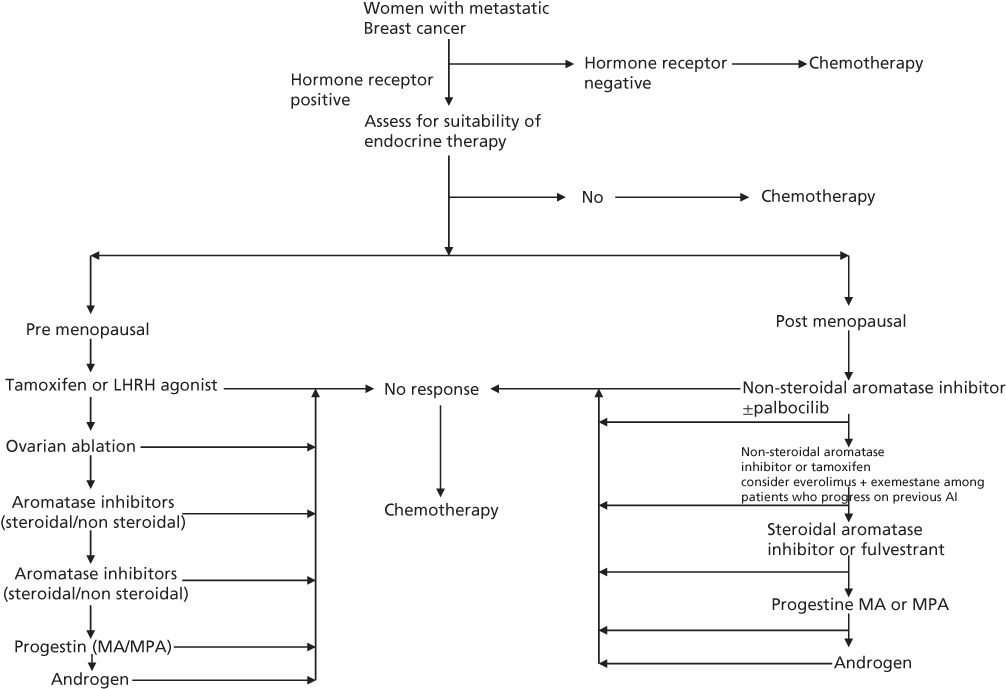
Figure 2 Algorithm for endocrine therapy among women with metastatic breast cancer.
Treatment of uterine cancer
Progestins are also used in the treatment of endometrial carcinoma.15 When diagnosed, adenocarcinoma of the uterus is cured by local therapy in 80% of cases. In the event of recurrence, exogenous progestin is an effective treatment in a significant fraction of cases: more than 30% of patients with recurrent disease demonstrate an objective response to exogenous progestins. ER and PR can be measured in these tumors, and the presence of these receptors correlates with differentiation of the tumor, prognosis for the patient, and response to progestins. The duration of response is not predicted by the presence of a receptor and varies from months to years. Tumors that lack ER and PR respond objectively to progestins in fewer than 10% of cases.15
Side effects and dosing of progestin
The most common adverse effect of progestin is weight gain, which occurs as a result of increased appetite and fluid retention. Its appetite-stimulating effect has frequently been used to treat cancer-induced cachexia. Other reported side effects include hot flashes, sweating, vaginal bleeding, nausea, dyspnea, thromboembolism, and rare cardiovascular events such as heart failure. Various dosing regimens of MA and MPA have been studied with a possible dose–response effect observed. The recommended dose of MA is 160 mg/day and that of MPA is at least 400–500 mg/day.16
Antiprogestins
Antiprogestins have wide and varied therapeutic applications including uses as contraceptives, to induce labor or to treat breast cancer, endometriosis, uterine leiomyomas, and meningiomas.17 The oldest and most widely used antiprogestin is RU 38486 or mifepristone that is a derivative of the 19-norprogestin norethindrone containing a dimethyl-aminophenol substituent at the 11β-position.11, 18 This compound is effectively absorbed orally and appears to bind with PR with high affinity and to effect altered coregulatory protein interaction after binding. It has been shown to have both antagonist and some agonist activity and is thus considered to be a PR modulator. Together with prostaglandins it is used for the termination of early pregnancy.18
Biology of estrogen production and action
A number of naturally occurring endogenous estrogens are produced in women with the most potent for both ER α- and β-mediated actions being estradiol followed by estrone and estriol. All three contain a phenolic A ring with a hydroxyl group at carbon 3 and a β-OH or ketone in position 17 of ring D with the phenolic A ring being the principle structural feature responsible for their selective high-affinity binding to both ERs.
The principle role of naturally occurring estrogens is to modulate cell growth by causing an increase in stimulatory growth factors [e.g., transforming growth factor-alpha (TGF-α)] and a decrease in inhibitory growth factors (e.g., TGF-β).19 These growth factors are thought to initiate or prevent progress through the cell cycle by interaction with their respective membrane receptors, with the regulatory mechanism functioning as an autocrine loop. There are also paracrine (cell–cell) influences of growth factors [e.g., insulin-like growth factor 1 (IGF-1)] that can play a role in modulating the replication of epithelial cells.
Biosynthetic pathway
The aromatase enzyme complex is located in the endoplasmic reticulum and consists of a cytochrome P450 hemoprotein (P450 AROM, aromatase) and the flavoprotein nicotinamide adenine dinucleotide phosphate (NADPH) that is common to most cell types and whose principle function is to donate electrons to cytochrome P450.20 The principle enzyme involved in the conversion of androstenedione to estrone in the estrogen biosynthetic pathway is aromatase, a product of the CYP 19 gene that encodes a polypeptide of 503 amino acids with a molecular weight of 55 kilodaltons (KDa). Aromatase catalyzes three separate steroid hydroxylations involved in the conversion of androstenedione to estrone. The first two reactions give rise to 19-hydroxy and 19-aldehyde structures, and the third, although still controversial, probably involves the C-19 methyl group with release of formic acid.21
Sites of production
A number of tissues have the capacity to express aromatase and hence synthesize estrogens, and these include the ovary, placenta, hypothalamus, liver, muscle, adipose tissue, and malignant breast tumor tissue22 (Figure 3).
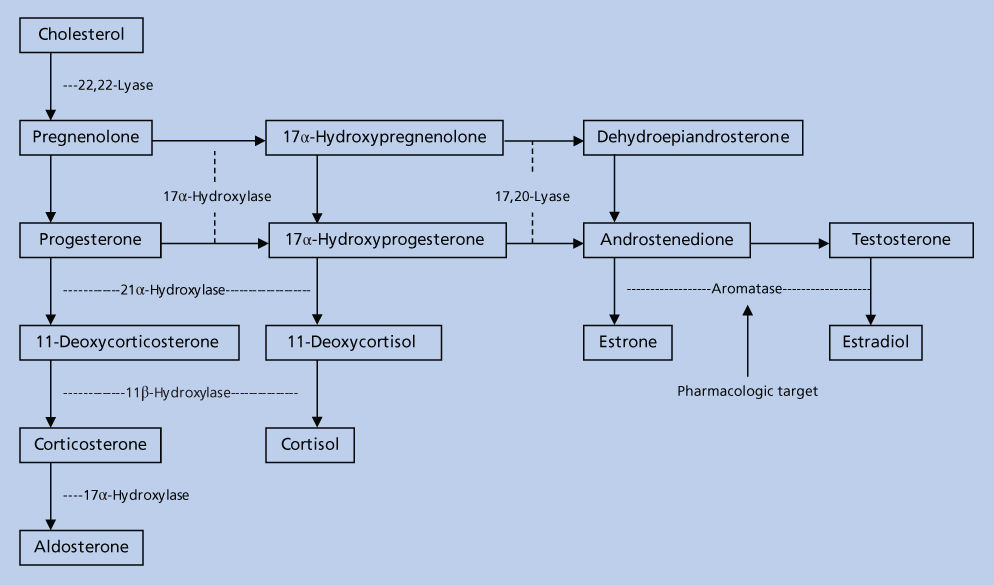
Figure 3 Sources of estrogen in postmenopausal women. The adrenal gland secretes androstenedione (A), which enters plasma and then tissue. Extraglandular and breast tumor tissues contain the enzymes necessary to convert A to estrone (E1) and to estradiol (E2) or to estrone sulfate (E1S). These steroids circulate in the plasma at levels indicated within the brackets and are expressed as picogram per milliliter.
In premenopausal women, the ovary is the most important site of aromatase and estrogen production. Luteinizing hormone (LH) controls production of androstenedione by the theca cell compartment, while follicle stimulating hormone (FSH) upregulates aromatase expression in granulose cells. Acting in concert, LH stimulates production of the substrate for aromatase, whereas FSH increases the amount of aromatase so that estradiol production can increase by 8- to 10-fold at the time of ovulation.
In postmenopausal women estrogen production takes place almost exclusively in extraglandular tissue. Androstenedione, produced primarily by the adrenal and, to a negligible extent, by the ovary, is converted to estrone by aromatase expressed in peripheral tissue such as adipose tissue23 and then subsequently converted to estradiol by 17-hydroxysteriod dehydrogenase. Through this pathway postmenopausal women produce approximately 100 mg of estrone per day, with higher levels observed in obese women.24 A fraction of estrone is also converted to estradiol to produce circulating plasma concentrations of approximately 10–20 pg/mL.
Estradiol levels in human breast tumor tissue are estimated to be 4–6 times these observed in plasma.25 Mechanisms by which such high levels are maintained have not been completely defined; however local production via the aromatase pathway is most likely involved with additional pathways involving steroid sulfatase, an enzyme known to hydrolyze estrone sulfate to estrone, having also been implicated.26
Mechanism of action
Circulating estrogens are bound to sex hormone binding globulins (SHBG) from which they dissociate and subsequently enter cells to exert their effects by binding to ERs located predominantly in the nucleus and bound to heat shock proteins (predominantly Hsp90) that stabilize them. The two ERs α- and β- are estrogen-dependent nuclear transcription factors, with different tissue distributions and transcriptional regulatory effects that are encoded by ESR1 and ESR2, respectively, that are located on separate chromosomes.27 ER α is expressed predominantly in the female reproductive tract including the uterus, vagina, and ovaries as well as the mammary gland, hypothalamus, endothelial cells, and vascular smooth muscles. ER β expression is found mainly in the prostate and ovaries with lower expression exhibited in the lung, brain, bone, and blood vessels. Coexpression of both ER α and β receptors are found in several tissues, the most notable being the mammary tissue where they form either homo- or heterodimers.
Binding of estrogen to the ERs results in a conformational change in the receptor leading to the release of ER from the stabilizing proteins. The estrogen–ER homodimeric complex then binds to a specific sequence of nucleotides called estrogen response elements (EREs) that are located in the promoter region of various genes. This binding interaction also involves a number of nuclear proteins, coregulators, as well as other components of the transcription machinery. Thus the genomic effects of estrogen are mainly the result of proteins synthesized from the regulation of transcription of a responsive gene. Two main modalities of treatment have been developed to treat estrogen-responsive breast cancers that either target preventing its production or inhibiting its interaction with ERs.
Carcinogenic effects
The major concern with the use of synthetic estrogens either alone or as a part of the preparation of oral contraceptives has been the development of cancer. Studies that reported the link between the intake of diethylstilbestrol (a synthetic estrogen) during the first trimester of pregnancy and the incidence of clear cell vaginal and cervical adenocarcinoma in later life of the offspring exposed in utero established for the first time that developmental exposure to estrogens was associated with an increase in human cancer.28, 29 Studies have also shown that unopposed estrogen as part of the HRT in postmenopausal women increased the risk of endometrial cancer by 5- to 15-fold30 with the increased risk prevented by the addition of a progestin.31
The relation of breast cancer risk and HRT in postmenopausal women has also been reported by two large trials. The Women’s Health Initiative (WHI) was a large prospective trial that randomized women to either placebo or HRT. The investigators reported an increase in total risk of breast cancer of 24% among women who took an estrogen–progestin combination and a decrease of 23% among women without a uterus who took estrogen only compared to women who took placebo.10, 32 In the extended poststopping phases of the WHI trials, the investigators further reported that some elevation in the risk of breast cancer risk still persisted in the cumulative follow-up period [HR 1.28, 95% CI (confidence interval) 1.11–1.48].33 The Million Women Study (MWS) was a large cohort study that reported an increased relative risk (RR) of invasive breast cancer among women who did and did not take HRT.34 Among women who took an estrogen–progestin combination, the increased RR of invasive breast cancer was 2 and that for women who took estrogen alone was reported as 1.3.
Epidemiological studies have also linked high levels of natural estrogens to the development of cancer. High levels of natural estrogens are observed in women who are overweight. Among postmenopausal women who had never received HRT in the WHI, those who were heavier (body mass index >31.1) had an elevated risk of breast cancer compared to slimmer women (body mass index <22.6) (RR = 2.52; 95% CI = 1.62–3.93).32 High levels of natural estrogens were also found to be associated with breast cancer risk in a case-cohort study involving women who had never received exogenous estrogens.35 The investigators reported that compared to women with the lowest levels of circulating estradiol, those with the highest levels (> or = 6.83 pmol/L or 1.9 pg/mL) had a RR of 3.6 (95% CI, 1.3–10.0).
Bone mineral density has also been shown to be a surrogate marker of estrogen exposure. A high endogenous estrogen concentration has been reported to be associated with greater bone mineral density in elderly women.36 Postmenopausal women with higher bone mineral densities have also been shown to have a higher incidence of breast cancer.37 Such studies serve to indicate the potential benefit of circulating estrogens as a surrogate marker of breast cancer risk. However its reliability as a surrogate marker is controversial due to the low baseline levels observed among postmenopausal women and the timing of circulating estrogen level measurement with relation to the menstrual cycle being important in premenopausal women. Regardless, enough evidence exists connecting estrogens to the development of hormone-responsive breast cancer that has spawned a number of prevention trials that have used agents targeted either at blocking the production of estrogens or its interaction with its receptor.
Selective estrogen receptor modulators and antiestrogens
The first indication of the role of hormones in the development of breast cancer occurred more than a century ago when in 1896, Beatson observed that remission could be induced by removal of the ovaries in a subset of breast cancer patients.38 Although not originally understood the observed effects occurred as a result of eliminating the primary source of estrogen in premenopausal women. This was confirmed in preclinical studies that demonstrated estradiol to promote proliferation of ER-positive breast cancer cells in culture39 and numerous epidemiological studies that have linked estrogens to breast cancer risk.32–37 With the realization of the important role estrogen played in the development and progression of breast cancer, two groups of drugs were developed to counteract the action of estrogens. The first group essentially prevented the interaction of estrogen to its receptor and included the selective estrogen receptor modulators (SERMs) and antiestrogens. SERMs including tamoxifen, raloxifene, and toremifene display unusual tissue-selective pharmacology having estrogen agonist properties in some tissues (bone, liver, and cardiovascular system), estrogen antagonist properties in other tissues (brain and breast), and mixed agonist/antagonist estrogen properties in the uterus. The antiestrogens which include fulvestrant are distinguished from SERMs in that they are uniformly estrogen antagonists. The second group of drugs blocks the production of estrogen by blocking the action of the aromatase enzyme and is known as aromatase inhibitors. In this section we will review the various SERMs and antiestrogens used in clinical practice for the treatment and prevention of hormone receptor-positive breast cancers (Figure 4).
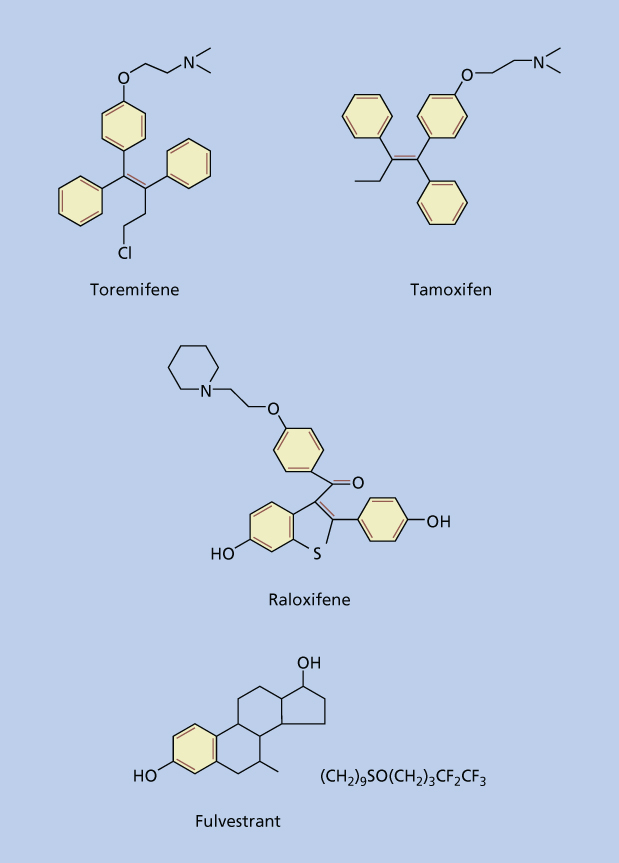
Figure 4 Structure of selective estrogen receptor modulators (SERMs) and antiestrogens.
Tamoxifen
Mode of action
Tamoxifen is a nonsteroidal triphenylethylene compound that exerts its effects by competitively inhibiting the binding of estradiol to ER thereby negating the stimulatory effects of estrogen causing the cell to be held at the G1 phase of the replication cycle.40 Tamoxifen is an estrogen antagonist in the breast and an estrogen agonist in the endometrium and bone, and it is this balance in biological properties that is key to the current strategies for the use of tamoxifen.
Clinical pharmacology
The high therapeutic index of tamoxifen has permitted wide variations in dosage with schedules and dosage of treatment varying depending on the country and its initial clinical trials that evaluated efficacy of this drug. Schedules of 10 mg twice daily or 20 mg once daily are recommended in the United States, although 10 mg three times daily and 20 mg twice daily have been used in other countries.
Tamoxifen is administered orally and is rapidly absorbed, achieving steady state serum levels within 4–6 weeks. It is metabolized in the liver by the cytochrome P450 isoform CYP2D6 and CYP3A4 into N-desmethyltamoxifen (major metabolite) and 4-hydroxytamoxifen (minor metabolite); both of which have the potential to be further metabolized to 4-hydroxy-N-desmethyltamoxifen (minor metabolite endoxifen) which have 30–100 times more affinity to the ER compared to tamoxifen itself.41 Tamoxifen has a long serum half-life of 7 days, and the metabolite N-desmethyltamoxifen has an even longer half-life of 14 days.42 These long serum half-lives are probably why a withdrawal response has not been routinely documented when tamoxifen therapy is discontinued. Currently no clinical cases of teratogenesis have been documented with tamoxifen. As the potential for teratogenicity is unclear, it is as such not recommended in pregnant women. Furthermore tamoxifen is known to cause ovarian stimulation in premenopausal women with ovulatory cycles, and thus women taking tamoxifen who are at risk of getting pregnant should be counseled about various contraceptive options.43
Tamoxifen in advanced breast cancer
Tamoxifen is an endocrine option for metastatic disease in postmenopausal women and those with ER-positive disease are more likely to benefit from this therapy.44 More recent data suggests that aromatase inhibitors are a better option as first-line treatment for this cohort of patients so long as resistance to the drug has not developed. This will be discussed in further detail in the section of “Aromatase inhibitors.”
Tamoxifen is an option for first-line endocrine therapy in premenopausal women with advanced breast cancer. A meta-analysis of individual patient data from four studies has demonstrated that tamoxifen produces a response rate and overall survival (OS) similar to what is seen after oophorectomy.45 However, with the development of effective luteinizing hormone-releasing hormone (LHRH) agonists such as goserelin (Zoladex), which acts to reduce ovarian steroidogenesis by preventing LH release from the pituitary gland, the combination of LHRH agonist and tamoxifen has become established as an effective therapeutic option, and the combination of which in meta-analysis has been shown to significantly improve progression free and OS compared to LHRH agonist alone.46 Hence recent guidelines have suggested that the use of LHRH agonists and tamoxifen alone or in combination are appropriate therapeutic options for premenopausal women with advanced metastatic hormone receptor-positive disease47 (Figure 3).
Tamoxifen in the adjuvant setting
Many randomized trials have addressed the question of tamoxifen efficacy in the adjuvant setting among women with early-stage breast cancer. An overview and meta-analysis of the results from 145,000 women with early-stage breast cancer who were randomized to 194 trials of adjuvant systemic therapy (chemotherapy and/or hormonal therapy) were recently updated by the Early Breast Cancer Trialists’ Collaborative Group (EBCTCG).48 The EBCTCG reported that 5 years of adjuvant tamoxifen among women with ER-positive disease resulted in reduction in the annual breast cancer death rate by 31% regardless of age, PR status, menopausal status, or use of chemotherapy with benefit persisting up to 15 years of follow-up. In this report 1 year of tamoxifen conferred little benefit, 5 years of tamoxifen was significantly more effective than 2 years, with long-term follow-up still required for assessing the benefit of more than 5 years of adjuvant tamoxifen treatment. A number of prospective trials have looked at the efficacy of tamoxifen beyond 5 years in the adjuvant setting (Table 1).49–53 The B-14 trial, a National Surgical Adjuvant Breast and Bowel Project (NSABP) trial, in which women with lymph node-negative ER-positive disease still in remission after receiving 5 years of tamoxifen were randomized to receive either placebo or more prolonged therapy with tamoxifen, did not show any advantage from prolonged tamoxifen treatment after 7 years of follow-up.49 Indeed longer duration of tamoxifen use was associated with shorter disease-free survival (DFS) compared to the group who had stopped taking tamoxifen after 5 years (78% vs 82%, p = 0.03). Recently long-term follow-up data of two large phase III trials that randomized women with early-stage breast cancer who had completed 5 years of tamoxifen to either another 5 years tamoxifen or stopping endocrine therapy have been reported.50, 51 In the Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) trial, 12894 women with early breast cancer were enrolled.50 Among the 6846 women who had ER-positive disease, the investigators reported a significant reduction in the risk of breast cancer recurrence (p = 0.002), breast cancer mortality (p = 0.01), and overall mortality (p = 0.01) favoring patients who were randomized to receive 10 years of tamoxifen. The investigators further reported that between years 5 and 14 of follow-up, 10 years of tamoxifen resulted in an absolute reduction in risk of breast cancer recurrence and mortality of 3.7% and 2.8%, respectively. However, between years 5 and 14 of follow-up, there was also an increased cumulative risk of endometrial cancer among patients allocated to 10 years of tamoxifen (3.1% vs 1.6%) which was associated with an absolute mortality increase of 0.2%. In the Adjuvant Tamoxifen Treatment Offer More (aTTom) trial, 6953 women with early breast cancer were enrolled.51 Similar to the ATLAS trial, the investigators reported reductions in breast cancer recurrence, breast cancer specific mortality, and overall mortality favoring patients who received 10 years of tamoxifen. The investigators observed that continuing tamoxifen beyond 5 years reduced breast cancer recurrence beginning year 7 and breast cancer mortality after year 10. In view of the evidence from these two clinical trials, current guidelines recommend consideration of 10 years of adjuvant tamoxifen.47 This should be considered after full discussion about potential associated increase in associated side effects.
Table 1 Efficacy data from trials comparing continuing tamoxifen beyond five years and stopping it
| Trial | Intervention | Risk of recurrence | Breast cancer specific mortality | Disease-free survival | Overall survival |
| ECOG53 (1996) | Continue vs stop | 15% vs 25% (p = 0.014 among ER + patients) | 8% vs 8.6% | 85% vs 73% (p = 0.10) | 86% vs 89% (p = 0.81 among ER+ patients) |
| NSABP-1449 (2001) | Continue vs stop | NR | NR | 78% vs 82% (p = 0.03) | 91% vs 94% (p = 0.07) |
| Scottish cancer trials breast group52 (2001) | Continue vs stop | 5.2% vs 7.1% | 23% vs 15% | 54% vs 61% (p = 0.15) | 59.5% vs 68% (p = 0.12) |
| ATLAS50 (2012) | Continue vs stop | RR = 0.81 (premenopausal) RR = 0.85 (postmenopausal) | 12.2% vs 15% (p = 0.01) during years 5–14 RR = 0.75 (p = 0.002) after year 10 | NR | NR |
| aTTom51 (2013) | Continue vs stop | 28% vs 32% (p = 0.003) | RR = 0.75 (p = 0.007) after year 10 | NR | 24.5 % vs 26.1% (p = 0.1) |
Tamoxifen and chemotherapy
The latest update of the EBCTCG revealed that addition of anthracyline-based polychemotherapy regimens was reported to be associated with annual reduction of mortality of 38% and 20% among women aged <50 years and 50–69 years, respectively.48 The question however is whether the addition of an endocrine agent such as tamoxifen could further add to this benefit among women with ER-positive breast tumors. In the EBCTCG among 3330 women with ER-positive or ER-unknown tumors, 28.1% of women who received only chemotherapy experienced a recurrence compared to 17.5% who received chemotherapy and 5 years of tamoxifen, with the difference being statistically significant.48 Similarly, among women with ER-positive or ER-unknown breast tumors, for those who were less than 50 years of age and received chemotherapy and tamoxifen, the EBCTCG reported a recurrence rate ratio of 0.64 (SE = 0.08) and annual breast cancer mortality ratio of 0.65 (SE = 0.10) compared to those who received tamoxifen alone. In addition similar trends were observed among women in the 50–69 years age group. Therefore chemotherapy alone is not enough among women with ER-positive tumors with the data clear that the addition of tamoxifen is important. The EBCTCG also explored the question about the sequence of chemotherapy and tamoxifen. Among women 50–69 years of age, a recurrence rate ratio and annual breast cancer mortality rate ratio of 0.80 (SE = 0.03) and 0.90 (SE = 0.03) was reported, respectively, among women treated with chemotherapy and tamoxifen compared to those who received tamoxifen alone. A recurrence rate ratio and annual breast cancer mortality rate ratio of 0.77 (SE = 0.08) and 0.80 (SE = 0.10) was reported, respectively, among women treated with chemotherapy followed by tamoxifen compared to those who received tamoxifen alone.48 The question about sequence of therapy was also explored by the Southwest Oncology Study Group (SWOG) 8814 study that reported improved disease-free and OS outcomes when tamoxifen was given sequentially following CAF (cyclophosphamide, doxorubicin, and 5-fluorouracil) compared with concurrent administration or tamoxifen alone.54 Taken together the data are suggestive that the sequential hormonal therapy with chemotherapy may be a better a better option.
Prevention of breast cancer
Observations that long-term tamoxifen therapy reduced the incidence and risk of contralateral breast cancer in women with early-stage breast cancer fueled interest in exploring the effect of tamoxifen in preventing the occurrence of breast cancer. An overview of the main outcomes from the five main breast cancer prevention trials, covering more than 28,000 patients, has shown that tamoxifen produced a 38% reduction in breast cancer incidence (p < 0.0001).55 There was no effect on ER-negative disease (p = 0.21), but ER-positive cancers were reduced by 48% (p < 0.0001). However, endometrial cancer rates were increased (consensus RR 2.4; p = 0.0005) in patients receiving preventive tamoxifen, as were venous thromboembolic events (relative risk 1.9; p < 0.0001). Thus when considering using tamoxifen as a preventive agent, it will be important to balance the risks of developing complications to that of developing breast cancer, and as such the unrestricted use of tamoxifen among young women of reproductive age should be used with caution. An exception to this is when tamoxifen is used for the reduction of breast cancer risk in high-risk women, for which it is the first medicine to be approved by the US Food and Drug Administration. Thus use of tamoxifen in preventive setting should be individualized to a woman’s risk of developing breast cancer. As such, women with a prior history of ductal carcinoma in situ, lobular carcinoma in situ, atypical hyperplasia, or those with a deleterious mutation of BRCA 1 or 2 are considered to be at higher risk of developing breast cancer and represent an ideal cohort to target where benefit outweighs risk of adverse events associated with tamoxifen.56–58 The side effects associated with tamoxifen meant that it was important to evaluate the action of other agents such as aromatase inhibitors in the preventive setting. The results of studies evaluating aromatase inhibitors as preventive agents will be discussed under the section titled “Aromatase inhibitors.”
Raloxifene
Raloxifene is a benzothiophene second-generation SERM that is FDA approved for the treatment and prevention of osteoporosis and for the reduction of risk of invasive breast carcinoma in postmenopausal women with either osteoporosis or who are at high risk for invasive breast cancer, respectively.59–62 This agent has no significant antitumor activity in the metastatic setting and as such is not approved for the treatment of advanced metastatic breast cancer. Four large prospective trials have reported on the efficacy of raloxifene as a chemopreventive agent for breast cancer. The Multiple Outcomes of Raloxifene Evaluation (MORE) trial that randomized 7705 postmenopausal women with osteoporosis to receive either raloxifene or placebo, whose primary end point was development of a fracture, was the first major trial to suggest raloxifene as a potential agent for chemoprevention of breast cancer.59 In this trial, following 4 years of treatment, raloxifene reduced the risk of ER-positive invasive breast cancer by 84% (RR 0.16, 95% CI 0.09, 0.30). The Continuing Outcomes Relevant to Evista (CORE) trial was an extension of the MORE trial to examine the effect of 4 additional years of raloxifene therapy on the incidence of invasive breast cancer in women enrolled in MORE who agreed to continue on the trial.60 Combining the 8 years of follow-up of both the MORE and CORE trials, the investigators reported that the incidences of invasive breast cancer overall and ER-positive invasive breast cancer were reduced by 66% (HR = 0.34; 95% CI = 0.22–0.50) and 76% (HR = 0.24; 95% CI = 0.15–0.40), respectively, in the raloxifene group compared with the placebo group. The goal of the Raloxifene Use for the Heart (RUTH) trial was to investigate the effect of raloxifene on the incidence of coronary events and breast cancer in 10,101 postmenopausal women.61 The investigators found reductions in breast cancer similar in size to that seen for tamoxifen in other studies. The NSABP P-2 trial is a prospective, double-blinded, randomized clinical trial that compared the efficacy and safety of tamoxifen with raloxifene on the risk of developing invasive breast cancer in a cohort of 19,747 postmenopausal women.62 The investigators reported similar efficacy of tamoxifen compared to raloxifene in reducing the risk of invasive breast cancer (RR = 1.02; 95% CI 0.82–1.28). In terms of side effects, compared to tamoxifen, raloxifene had fewer gynecological and thromboembolic events. Interestingly raloxifene reduced the risk of invasive breast cancer but had no effect on the incidence of ductal carcinoma in situ.
Lasofoxifene
Lasofoxifene is a third-generation nonsteroidal SERM that has been shown to selectively bind to both ER α- and β with a high affinity and a median inhibitor concentration that is 10-fold higher than reported for raloxifene and tamoxifen. Furthermore it has demonstrated improved oral bioavailability when compared to other SERMs due to its increased resistance to intestinal wall glucuronidation.63 Among postmenopausal women lasofoxifene has been shown to decrease bone loss, bone resporption, and low density lipoprotein (LDL) cholesterol. In a prospective randomized clinical trial, 8556 postmenopausal women who had a bone mineral density T score of −2.5 or less were randomly assigned to receive lasofoxifene or placebo for 5 years.64 The investigators reported that compared to placebo, use of lasofoxifene among postmenopausal women with osteoporosis was associated with a reduced risk of ER-positive breast cancer, coronary heart disease, stroke, and nonvertebral and vertebral fractures. However it was also reported to be associated with an increased risk of venous thromboembolic events.
Toremifene
Toremifene (Fareston) is a structural derivative of tamoxifen with similar antiestrogenic and estrogenic properties demonstrated in laboratory animals. In general, toremifene is highly protein bound, which could explain its long serum half-life. Toremifene is less potent than tamoxifen, and, consequently, clinical studies have evaluated doses of toremifene up to 240 mg/day. Toremifene is cross-resistant with tamoxifen, but clinical trials have shown that it exhibits a similar efficacy and side effect profile to tamoxifen and so may be used as an alternative to treat advanced breast cancer.65, 66 At this time there is insufficient data to recommend its use in the adjuvant setting.
Trilostane
Trilostane (Modrenal) is an antiadrenal drug that is usually used for short-term adrenal suppression in the treatment of Cushing’s syndrome. However, trilostane’s ability to modify the binding of estrogen to the ER has prompted interest in its potential to block breast tumor cell proliferation. A meta-analysis of several small studies investigating the use of trilostane in postmenopausal women with advanced breast cancer reported clinical benefit with this agent, and further trials are needed to evaluate its worth as an endocrine therapy for advanced breast cancer.67
Fulvestrant
Fulvestrant (Faslodex) is an antiestrogen with no agonist properties and unlike tamoxifen has the following mechanism of action: it binds, blocks, and increases degradation of ER protein, leading to an inhibition of estrogen signaling through the ER together with dramatic loss of cellular ER levels and is also associated with a significant reduction in PgR expression.68, 69 A prospective combined analysis of two phase III trials comparing fulvestrant (250 mg IM injection once monthly) to anastrozole in a cohort of postmenopausal women with advanced breast cancer who had progressed on prior tamoxifen therapy indicated that, after a median follow-up of 15.1 months, fulvestrant was at least as effective as anastrozole [median times to progression (TTP) were 5.5 months vs 4.1 months] with a similar and acceptable adverse event profile.70 Subsequently fulvestrant (at a dose of 250 mg IM) as first-line treatment of advanced breast cancer was compared to tamoxifen among postmenopausal women (whose tumors were either hormone receptor positive or hormone receptor unknown) in a randomized clinical trial.71 At a median follow-up of 14.5 months in the overall population, noninferiority of fulvestrant compared to tamoxifen could not be demonstrated. In a preplanned subgroup analysis focusing on the cohort of patients with hormone receptor-positive disease, fulvestrant was observed to have a similar efficacy to tamoxifen. In the setting of a phase III, randomized clinical trial fulvestrant (at a dose of 250 mg IM) has also been compared to exemestane for the treatment of postmenopausal women with hormone receptor-positive advanced breast cancer who had progressed or recurred on a nonsteroidal aromatase inhibitor.72 In this study overall response rate (7.4% vs 6.7%; p = 0.736) and time to treatment progression (3.7 months in both groups) were similar between the fulvestrant and exemestane groups suggesting that fulvestrant was no more effective than a steroidal aromatase inhibitor among women who had progressed on a nonsteroidal aromatase inhibitor. Since fulvestrant can take 3–6 months to reach steady state plasma levels at the 250 mg/month dose (previously approved dosing schedule), clinical trials have evaluated the standard dosing regimen to a loading dosing schedule. In a phase III double-blinded multicenter study, postmenopausal women with ER-positive advanced breast cancer who had progressed on prior endocrine therapy were assigned to receive IM monthly fulvestrant at a dose of 500 mg or 250 mg.73 The investigators reported a significantly longer DFS with the 500 mg dosing schedule (HR 0.8, 95% CI 0.68–0.94, p = 0.006) with no associated increase in toxicity. Based on these results fulvestrant is approved at a dose of 500 mg a month for the treatment of hormone receptor-positive metastatic breast cancer in postmenopausal who had progressed on prior antiestrogen therapy. As the 500 mg dosing schedule was found to be superior to the 250 mg dosing schedule, trials are underway investigating whether the use of the new approved dose of fulvestrant is superior to aromatase inhibitors in the treatment of advanced hormone receptor-positive breast cancer in the first-line setting. In an open-labeled randomized phase II trial, postmenopausal women with advanced breast cancer were randomized in the first-line setting to either 500 mg of fulvestrant or anastrozole.74 The investigators reported a significantly longer time to progression of disease with fulvestrant compared to anastrozole. Updated results of this analysis have recently been presented with the investigators further reporting a significant improvement in OS with 500 mg of fulvestrant compared to anastrozole.75 Phase III trials are currently ongoing to confirm these results (FALCON trial).
Side effects of SERMs and antiestrogens
Side effects related to the SERMs and antiestrogens develop mainly as a result of the blockade of the stimulatory function of estrogen on a variety of tissues. The most frequent side effects encountered are hot flashes, night sweats, and vaginal dryness similar to that seen in women undergoing menopause. Other less frequent but important side effects pertain to the bone, blood vessels, and carcinogenic effects.
Osteoporosis
Estrogen is important in maintaining bone health in premenopausal women with HRT and is often recommended to prevent the development of osteoporosis in postmenopausal women. Long-term administration of an antiestrogen has the potential to cause premature osteoporosis in premenopausal women. However due to the partial estrogen agonist function of SERMs, clinical studies have shown tamoxifen therapy to not be associated with a reduction of bone density,76 and raloxifene is an approved treatment for osteoporosis.69
Coronary heart disease
Estrogen lowers LDL cholesterol levels and raises high-density lipoprotein (HDL) cholesterol levels, and thus prolonged administration of an antiestrogen could produce a population at risk of premature coronary heart disease. However, the estrogen-like effects of tamoxifen have been shown to lower the circulating levels of cholesterol in female patients77, 78 with clinical studies reporting tamoxifen to be associated with either a significant or trend in reduction of risk of coronary heart disease.79 Raloxifene has also been shown to reduce serum cholesterol levels80: however it has not been shown in a large randomized clinical trial to reduce the risk of coronary heart disease.61
Thromboembolism
A number of studies have demonstrated an association between the use of tamoxifen and subsequent thromboembolic episodes in both the treatment and preventive setting.55, 81 This is comparable with increases noted with HRT or raloxifene81 Patients with a known history of thromboembolic disorders should be carefully evaluated before a decision is made to use long-term tamoxifen therapy.
Endometrial tumors
Research has demonstrated that increases in endometrial thickness, hyperplasia, and fibroids may follow treatment with tamoxifen.82 Endometrial thickening is associated with the stromal component of the uterus rather than the epithelial component.83 Clinical trials evaluating the efficacy of tamoxifen in the treatment and prevention of breast cancer have demonstrated an increased risk of endometrial tumors including carcinomas and to a smaller extent sarcomas.84, 85 Endometrial carcinoma that develops on tamoxifen therapy is not of high grade and as such is not associated with poor prognosis, while endometrial sarcomas are generally associated with a poorer prognosis, seemingly because of less favorable histology and higher stage.86 Thus when monitoring patients on tamoxifen treatment, all cases of persistent vaginal bleeding should be followed up with a gynecological examination and an endometrial biopsy. It is important to note that this increased risk is restricted to postmenopausal women with premenopausal women not at risk of an increase in endometrial cancer. When raloxifene was directly compared to tamoxifen in the prevention of breast cancer (STAR trial), 36 cases of endometrial cancer were observed in the tamoxifen group compared to 23 cases in the raloxifene group (RR, 0.62; 95% CI, 0.35–1.08).62 In the ATLAS and aTTom trials that evaluated the efficacy of 5 years of tamoxifen to 10 years of tamoxifen in the adjuvant setting, the investigators reported that longer duration of tamoxifen was associated with increased incidence of endometrial cancer and associated increase risk of mortality from endometrial cancer.50, 51 The risk associated with tamoxifen usage appears confined to the duration of its administration. In the randomized International Breast Cancer Intervention Study (IBIS-I) (a prevention study that randomized patients to tamoxifen vs placebo), the rates of side effects pertaining to tamoxifen including deep vein thrombosis, pulmonary embolism, and endometrial cancer was higher in the tamoxifen arm of the study compared to the placebo arm during the active treatment period of the study but not in the subsequent years posttamoxifen treatment.87
Other side effects
Antiestrogens and SERMs have also been associated with ophthalmic side effects such as cataracts and retinal changes.88, 89 Preclinical studies have also demonstrated tamoxifen to cause carcinogenesis in the liver; however an increase in human hepatocellular carcinoma has not been demonstrated.90
Aromatase inhibitors
In contrast to SERMs and antiestrogens, aromatase inhibitors work by blocking the enzyme complex responsible for the final step in estrogen biosynthetic pathway and is thereby essentially preventing the production of the ER substrate. Moreover, unlike tamoxifen, aromatase inhibitors have no partial estrogen agonist function. Despite the ovaries being a rich source of aromatase, aromatase inhibitors are unable to sufficiently suppress ovarian estrogen production to postmenopausal levels which may be due to compensatory rise in gonadotrophins which maintains adequate estrogen production, despite the presence of the inhibitor. In contrast aromatase inhibitors have been shown to adequately suppress estrogen production in postmenopausal women.
Aromatase inhibitors are classified into first, second, and third-generation aromatase inhibitors according to the specificity and potency with which they inhibit the aromatase enzyme (Table 2). They are further subclassified according to their mechanism of action into steroidal (irreversible, type I) and nonsteroidal (reversible, type II) inhibitors. Type 1 inhibitors, including formestane and exemestane, function by irreversibly inhibiting the aromatase enzyme by covalently binding to it, resulting in permanent inactivation that persists even after discontinuation of the drug until the peripheral tissues synthesize new enzymes. Type II inhibitors, including anastrozole, letrozole, and fadrozole, in contrast bind reversibly to the active site of the aromatase enzyme and prevent product formation only as long as the inhibitor occupies the catalytic site. In this section we will focus on the newer third-generation aromatase inhibitors letrozole, anastrozole, and exemestane that are routinely used in clinical practice today. These aromatase inhibitors have challenged tamoxifen as the gold standard and are now the preferred first-line treatment of postmenopausal women with hormone-responsive breast cancer in either the early or advanced setting, whenever there is a moderate or high risk of relapse.
Table 2 Structure and classification of representative aromatase inhibitors
| Generation | Steroidal irreversible type I inhibitors | Nonsteroidal reversible type II inhibitors |
First generation | Aminoglutethimide | |
Second generation  | Formestane | Fadrozole |
Third generation | Exemestane | Anastrozole Letrozole Vorozole |
Compounds are shown in approximate order of increasing specificity and potency of aromatase inhibition.
First generation
Aminoglutethimide, a derivative of the sedative agent glutethimide, was initially introduced as an inhibitor of cytochrome P450 N-mediated steroid hydroxylations.91 The effects of this compound, however, are rather nonspecific, because the drug affects a number of hydroxylation steps in the metabolic conversion of cholesterol to active steroid products, and, overall, the use of aminoglutethimide plus glucocorticoid in women with breast cancer produces results similar to those expected from other forms of endocrine therapy. Side effects observed with standard doses of aminoglutethimide (1000 mg/day) include drug rash, fever, and lethargy.91 With the development of more selective second and third-generation aromatase inhibitors, aminoglutethimide is now rarely used for the treatment of breast cancer
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


