Fig. 5.1
Diagrammatic representation of the hypothalamic-pituitary axis
Table 5.1
Anterior pituitary hormones and major hypothalamic regulatory factors
Pituitary hormone | Hypothalamic factor | Effect |
|---|---|---|
Growth hormone | Growth hormone-releasing hormone | + |
Somatostatin | − | |
Prolactin | Dopamine | − |
Luteinizing hormone | Gonadotropin-releasing hormone | + |
Follicle-stimulating hormone | Gonadotropin-releasing hormone | + |
Thyroid-stimulating hormone | Thyroid-releasing hormone | + |
Somatostatin | − | |
Adrenocorticotropin | Corticotropin-releasing hormone | + |
Vasopressin | + |
5.2.1.1 Growth Hormone
Growth hormone (GH) is a 191-amino-acid polypeptide hormone synthesized and secreted by the somatotrophs in the anterior pituitary gland in response to hypothalamic releasing hormones, primarily GH-releasing hormone (GHRH). In addition, ghrelin secretion from the stomach during fasting also contributes to GH secretion [51]. GHRH levels are usually steady, while somatostatin secretion is interrupted intermittently. Somatostatin inhibits GH release but paradoxically contributes to synthesis of GH in the pituitary [12]. When somatostatin concentrations decrease, the tonic concentration of GHRH causes release of GH into the systemic circulation. Factors such as neuropeptide Y, leptin, galanin, and ghrelin also affect GH secretion. In healthy children and adults, GH secretion is pulsatile, particularly during sleep, with two to six pulses per night [61]. In adolescents, additional pulses occur during the day, and the pulses have higher peaks than those seen in children and adults (Fig. 5.2a).
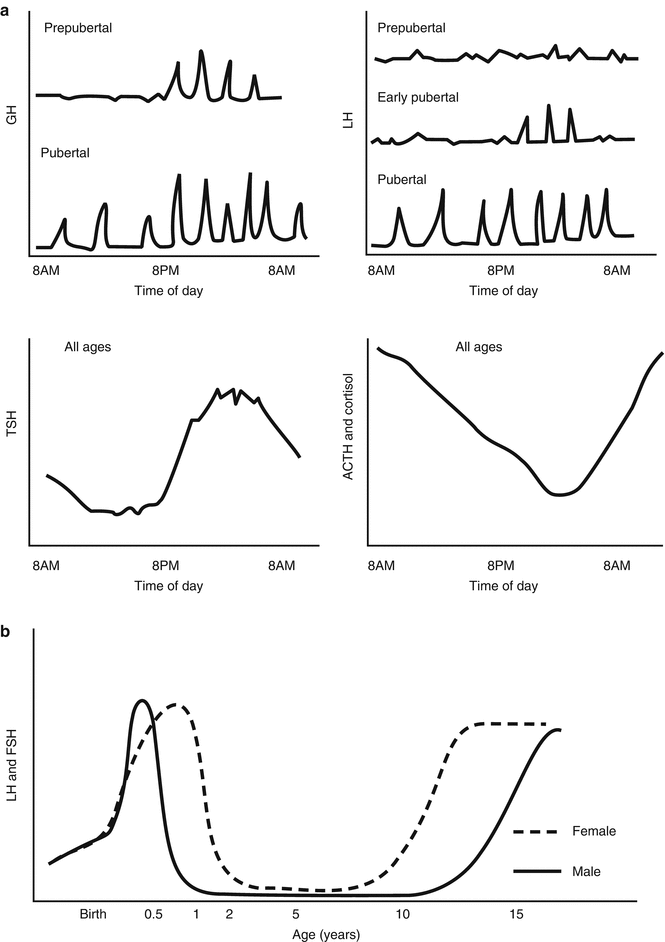
Fig. 5.2
Typical daily pattern of hormone secretion and changes with pubertal status and time of day in normal individuals (a) growth hormone (GH), luteinizing hormone (LH), thyroid-stimulating hormone (TSH), and adrenocorticotropin (ACTH) and cortisol secretion. (b) Normal changes in LH and FSH levels from infancy to adolescence
Circulating serum GH stimulates production of insulin-like growth factor I (IGF-I) in all tissues. IGF-I mediates GH effects on growth, bone mineralization, and body composition (decreased truncal fat deposition, increased lean muscle mass) [78]. IGF-I is bound to IGF-binding proteins such as IGFBP3 and is transported in the blood. IGF-I and IGFBP3 concentrations are stable during the day, and each reflects the integrated concentration of secreted GH.
5.2.1.2 Gonadotropins
Luteinizing hormone (LH) and follicle-stimulating hormone (FSH) are glycoprotein hormones both stored in the same cells in the anterior pituitary. Their overall patterns of secretion vary according to the age and gender of the person. The pituitary gland produces and secretes LH and FSH in a pulsatile manner in response to episodic release of GnRH from the hypothalamus (Fig. 5.2a). The hypothalamic stimulus is actively inhibited between 6 months of age and the usual age of onset of puberty (Fig. 5.2b). This inhibition can be disturbed by tumor mass, cranial surgery, or irradiation, thereby resulting in precocious puberty in children. In men, LH stimulates testosterone production in Leydig cells of the testes; normal spermatogenesis requires both LH and FSH. In women, FSH stimulates production of estrogen and LH stimulates production of progesterone in the ovary. The LH surge near the end of the follicular phase of the menstrual cycle is necessary to stimulate ovulation. Development of the ovarian follicles is largely under FSH control, and secretion of estrogen from follicles is dependent on both FSH and LH.
5.2.1.3 Thyroid-Stimulating Hormone
Thyrotropin, or thyroid-stimulating hormone (TSH), is a glycoprotein hormone synthesized in the anterior pituitary. Secretion of TSH is stimulated by TSH-releasing hormone (TRH) and inhibited by somatostatin and dopamine secreted from the hypothalamus. In persons older than 12 months of age, there is a circadian pattern to TSH release. TSH concentration is low after 1000 h and in the afternoon, rises dramatically (surges) after 1900 h, and reaches highest concentrations between 2200 and 0400 h (Fig. 5.2a) [58]. Thus, at least one third of the trophic influence of TSH on the thyroid gland occurs at night. TRH is necessary for TSH synthesis, posttranslational glycosylation, and secretion of a fully bioactive TSH molecule from the pituitary. Altered TSH glycosylation, resulting in altered bioactivity, is seen in mixed hypothyroidism (central hypothyroidism with mild TSH elevation [5–15 mU/L]) [34, 56].
TSH stimulates the thyroid gland to produce thyroxine (T4) and triiodothyronine (T3). T4 and T3 circulate in the blood stream bound to thyroxine-binding globulin and albumin; only small amounts are free or unbound. Free T4 undergoes intracellular deiodination to form free T3, which interacts with DNA in the cell nucleus to influence cellular mRNA and protein synthesis. Free T4 provides negative feedback at the hypothalamus and pituitary to modulate the secretion of TRH and TSH.
5.2.1.4 Adrenocorticotropin
Adrenocorticotropin (ACTH) is a 39-amino-acid peptide hormone processed in the corticotrophs from a large precursor molecule, pro-opiomelanocortin. In healthy individuals, hypothalamic corticotrophin-releasing hormone and vasopressin released in two or three synchronous pulses per hour synergistically stimulate secretion of ACTH from the pituitary [14]. ACTH secretion is pulsatile and varies throughout the day; it peaks before the person awakens in the morning (Fig. 5.2a), increases with stress, and is inhibited by glucocorticoid medications. Since cortisol secretion is regulated by ACTH, the pattern of cortisol secretion is similar to that of secretion of ACTH. In addition to the negative feedback of glucocorticoids, ACTH inhibits its own secretion (short-loop feedback).
5.2.1.5 Prolactin
Prolactin (PRL) is a 198-amino-acid polypeptide hormone synthesized and secreted from lactotrophs of the anterior pituitary. A precursor molecule is also secreted and can constitute as much as 10–20 % of the PRL immunoreactivity in the plasma of healthy persons. Hypothalamic control of PRL secretion (primarily through dopamine release) is different than that of the other pituitary hormones in that the hypothalamus inhibits secretion of PRL rather than stimulating it. Thus, elevated PRL levels can be a useful marker of hypothalamic disorders that leave the pituitary intact.
5.2.1.6 Antidiuretic Hormone
Antidiuretic hormone (ADH) or vasopressin is a peptide hormone synthesized in the hypothalamic neurohypophyseal neurons, transported through the pituitary stalk in long axons, and released from the nerve terminals in the posterior pituitary. ADH is secreted in response to reduced plasma volume and increased plasma osmolality. In normal individuals, ADH secretion is increased when there is no fluid intake, such as during sleep or in dehydration.
5.2.2 Injury of the Hypothalamic-Pituitary Axis in Patients with Cancer
The hypothalamic-pituitary axis (HPA) is vulnerable to damage by certain tumors, surgical trauma, irradiation, chemotherapy, and other common risk factors (Table 5.2) [18, 57]. Patients with tumors in the area of the HPA (e.g., craniopharyngioma or hypothalamic/chiasmatic tumor) are at particular risk for neuroendocrinopathy [23, 42]. Many HPA injuries are attributable to damage caused by radiation therapy (RT) [57]. However, the occurrence of pre-RT neuroendocrinopathies in pediatric patients with brain tumors is high. Of 68 pediatric patients in one study [44], 45 (66 %) showed evidence of neuroendocrinopathy before RT, including 15 of 32 patients with tumors in the posterior fossa not adjacent to the HPA. Seventeen of the 45 patients (38 %) had abnormality in GH, 19 (43 %) in TSH, 10 (22 %) in ACTH, and 6 (13 %) in gonadotropin. In addition, patients who receive chemotherapy alone [with no history of RT or central nervous system (CNS) tumor] may also be at risk for neuroendocrinopathy. Of 31 such patients referred after chemotherapy for evaluation of altered growth and development, 48 % had GH deficiency, 52 % had central hypothyroidism, and 32 % had pubertal abnormalities [62].
Table 5.2
Risk factors for endocrine disorders, diagnostic studies, and treatment options
Disorder | Highest risk | Diagnostic studiesa | Treatment optionsa |
|---|---|---|---|
GH deficiency | ≥18 Gy CRT Pretransplant CRT TBI Young age Tumor near HPA Hydrocephalus | IGF-I, IGFBP-3 Bone age radiograph GH stimulation tests | Recombinant GH (SC) GnRH agonist (IM) (If pubertal maturity too advanced for height) |
Gonadotropin deficiency | ≥30 Gy CRT Tumor near HPA | LH, FSH, AMH, inhibin, estradiol, or testosterone (4–8 AM) Bone age GnRH stimulation test | Estrogen/progestin (O or T) (female) Testosterone (IM or T) (male) |
Precocious puberty | 18–24 Gy CRT Female Young age Tumor near HPA | LH, FSH, estradiol, or testosterone (4–8 AM) Bone age Pelvic ultrasound (female) ± GnRH stimulation test ± GH stimulation test | GnRH agonist (IM) |
TSH deficiency | ≥30 Gy CRT TBI Tumor near HPA Hydrocephalus | Free T4, TSH (8 AM) AM-PM TSH ratio Nocturnal TSH surge | L-thyroxine (O) |
ACTH deficiency | ≥30 Gy CRT Tumor near HPA Hydrocephalus | Cortisol (8 AM) Low-dose ACTH stimulation test | Hydrocortisone (O) Stress dosing (O, IM, or IV) |
Hyperprolactinemia | ≥50 Gy CRT Tumor near HPA | Prolactin | Dopamine agonists (O) |
Diabetes insipidus | Histiocytosis Germinomas Tumor or tumor-related cysts near HPA | Simultaneous serum and urine osmolarity after 8–12 h without fluid intake Water deprivation test | Desmopressin (O) DDAVP (SC or IN) |
Osteopenia | Low GH, TSH, or LH/FSH High prolactin Low vitamin D intake | DXA or quantitative CT 25OH-vitamin D level | Calcium + vitamin D (O) ± Bisphosphonates (O or IV) |
Hypothalamic obesity | Young age (<6 years) ≥50 Gy (hypothalamus) Tumor near HPA | Fasting insulin and glucose Oral glucose tolerance test with insulin levels | Diet and exercise Ritalin or dexedrine (O) Metformin (O) (monitor for hypoglycemia) Octreotide (SC) Bariatric surgery |
GH deficiency is often the first hypothalamic-pituitary deficiency to emerge after injury to the HPA, followed by deficiencies of gonadotropin, TSH, and ACTH [18, 70]; however, these deficiencies can develop in any order [44, 59, 71]. Although the most common neuroendocrinologic abnormality in survivors of childhood cancer is GH deficiency (see Sect. 5.3.1), hypothyroidism is at least as prevalent when sensitive testing methods are used (see Sect. 5.3.4) [59]. The next most common alterations are in pubertal timing (early, rapid, precocious, delayed, or absent) (see Sects. 5.3.2 and 5.3.3). ACTH deficiency, though less common than the other disorders, has more serious consequences if it is not detected (see Sect. 5.3.5). Diabetes insipidus rarely develops after chemotherapy or irradiation, but commonly occurs after surgery in the hypothalamic-pituitary area or in association with histiocytosis or germinoma (see Sect. 5.3.7). Hypothalamic injury resulting from tumor, surgery, or irradiation can result in unrelenting weight gain, termed hypothalamic obesity (see Sect. 5.3.9). Finally, osteopenia may result from hypothalamic-pituitary deficiency, particularly GH deficiency, hypothyroidism, and hypogonadism (see Sect. 5.3.8).
5.2.3 Contribution of Radiation to Hypothalamic-Pituitary Axis Injury
Radiation therapy (RT) is a significant contributor to neuroendocrine complications observed after treatment for CNS tumors, CNS preventative therapy for leukemia, and following total body irradiation. Similar complications are observed when the HPA is incidentally irradiated in the treatment of nasopharyngeal cancer, retinoblastoma, Hodgkin disease with involvement of Waldeyer’s ring, and pediatric sarcomas of the head and neck (e.g., parameningeal and orbital rhabdomyosarcoma) (Fig. 5.3). The incidence and time to onset of neuroendocrine sequelae after RT are difficult to predict because of other contributors to HPA dysfunction that may coincide temporally with the administration of RT. A notable example is hydrocephalus which can cause mass effect in the region of the anterior third ventricle and generalized diminished blood flow to sensitive regions of the brain. In one study, 59 children with infratentorial ependymoma underwent provocative testing for GH, thyroid hormone, and ACTH secretion abnormality prior to RT [43]. Abnormal testing was observed in 27 patients (46 %) with 30 % of the 59 having abnormality in GH secretion. Serial measurements of ventricular size from the time of diagnosis to 1 year after RT were recorded and modeled to show that ventricular size at the time of diagnosis could be used to predict pre-irradiation endocrinopathy. Change in ventricular size over time could predict GH deficiency prior to irradiation (Fig. 5.4). This study demonstrated a relatively high rate of pre-irradiation endocrinopathy in a well-defined group, confirming another important tumor-related cause of endocrinopathy. Thus, management of hydrocephalus is important, particularly in children with posterior fossa tumors.
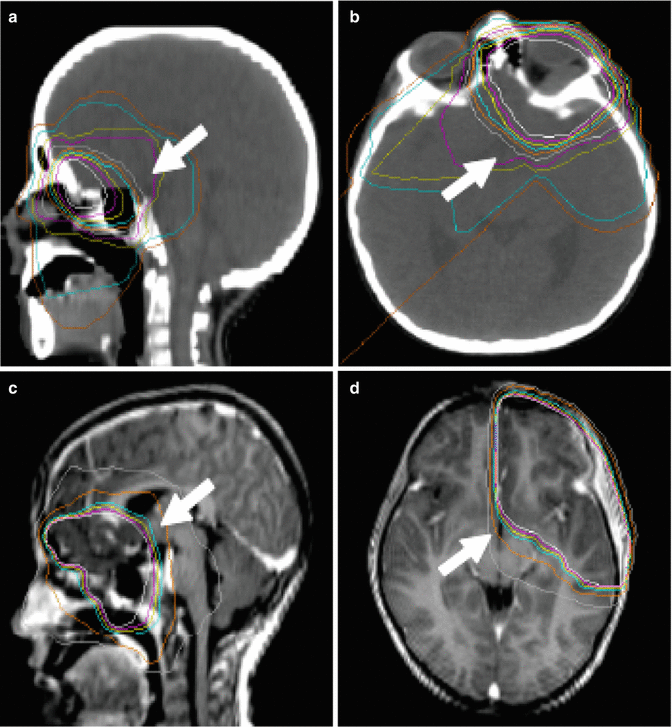
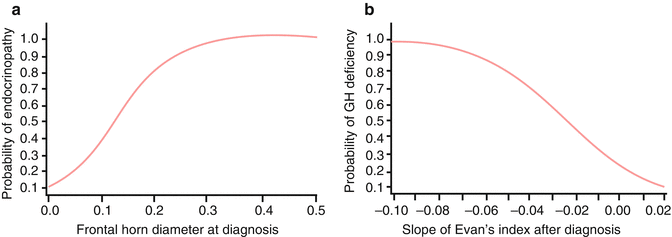
Fig. 5.3
Radiation dosimetry taken from the treatment of children with orbital (a, b) and infratemporal fossa rhabdomyosarcoma (c, d). The images illustrate cases in which the HPA is incidentally irradiated and may receive all or a portion of the prescription dose (arrow indicates location of the hypothalamus)
Fig. 5.4
The effect of hydrocephalus on pre-irradiation endocrinopathy in children with infratentorial ependymoma. (a) Probability of pre-irradiation endocrine deficiency based on frontal horn diameter measured at diagnosis. (b) Probability of pre-irradiation growth hormone (GH) deficiency based on change (slope) in the Evan’s index after diagnosis. The Evan’s index is the ratio of the distance between the most lateral extent of the frontal horns of the lateral ventricles and the width of the parietal brain at the same level
Many reports of neuroendocrine effects of RT have used generalized estimates of radiation dose under conditions where the dose to the HPA was relatively homogeneous and discrete [46]. Examples include patients treated with single-dose or fractionated TBI (8–14 Gy), cranial irradiation for leukemia (18 and 24 Gy) and tumors of the sellar or parasellar region in which the HPA was uniformly included in the volume of prescribed dose (>50 Gy) (Fig. 5.5). For other diseases, the HPA may have been located within the irradiated volume for part or all of the treatment or in the gradient of dose (dose fall off) experiencing only a fraction of the daily dose administered (Fig. 5.6). These circumstances make it difficult to assign a dose to the HPA and to determine the risk for late effects. When the patient is seen by the endocrinologist years after treatment, retrospective dose calculations may be difficult to perform. Newer radiation techniques employ three-dimensional imaging (computed tomography and magnetic resonance, CT and MR) in the planning process. The HPA and other normal tissues can be contoured on CT or MR data and the dose to the HPA calculated and reported more accurately [33]. This information can be correlated with objective measures of endocrine effects and can be used to predict incidence of specific endocrine effects. Already this type of data has been modeled to predict peak GH secretion after radiation therapy [46] and may in the future be used to optimize RT for children (Fig. 5.7, Table 5.3).
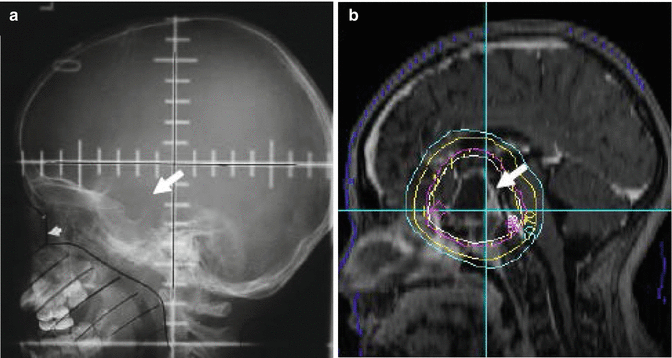
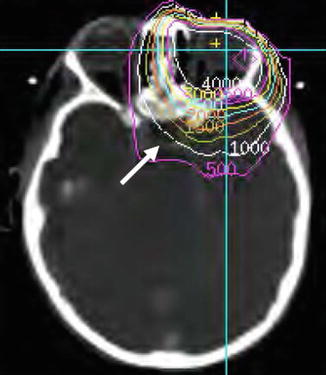
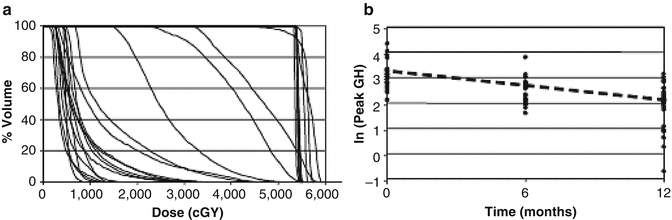
Fig. 5.5
Homogeneous irradiation of the HPA including (a) a traditional treatment portal used for cranial irradiation in ALL and (b) dosimetry from focal treatment of craniopharyngioma (arrow indicates location of the hypothalamus)
Fig. 5.6
Dosimetry for a typical patient treated with conventional radiation therapy (40 Gy). This example illustrates that the HPA receives only a portion of the total dose given to the primary tumor (arrow indicates location of the pituitary)
Fig. 5.7
HPA dose-volume data from patients treated with conformal radiation therapy. (a) Dose-volume curves represent the percent-volume of the hypothalamus receiving a specific dose. (b) Correlation with change in peak GH (ATT/L-dopa) measured before, 6 and 12 months after radiation therapy results in an estimating equation that can be used to predict GH deficiency up to 12 months after irradiation based on the volume (V) received dose over specified intervals. ln [peak GH] = 3.072 − (0.00058 × V 0–2,000 cGy + 0.00106 × V 2,000–4,000 cGy + 0.00156 × V 4,000–6,000 cGy) × time
Table 5.3
Probability of growth hormone deficiency according to hypothalamic radiation dose and according to time since irradiation
Hypothalamic radiation dose | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Time (Gy) | 15 | 20 | 25 | 30 | 35 | 40 | 45 | 50 | 55 | 60 |
12 months (%) | 17 | 19 | 22 | 25 | 28 | 31 | 34 | 38 | 42 | 45 |
36 months (%) | 26 | 37 | 48 | 59 | 70 | 79 | 86 | 91 | 95 | 97 |
60 months (%) | 39 | 57 | 75 | 87 | 95 | 98 | 99 | 100 | 100 | 100 |
In pediatric radiation oncology, reducing side effects of treatment is an important goal. Reducing side effects can be achieved by limiting CNS irradiation to those for whom indications are clear and benefits outweigh the risks. CNS irradiation has been eliminated from treatment of the majority of children with leukemia and a significant proportion of children with low-grade glioma who may be cured with surgery. However, CNS irradiation will remain a mainstay in treatment of most children with brain tumors. Incidental irradiation of the CNS will continue to be observed in children with ocular tumors or tumors of the head and neck. Increased awareness of the importance of the hypothalamus in radiation-related neuroendocrine sequelae, and use of three-dimensional imaging in planning treatment of these tumors, may lead to a reduction in late endocrine effects. Reducing risk for complications can also be achieved by delaying radiation therapy until the child is older or until chemotherapy has had a chance to shrink the tumor and reduce the field of radiation [5, 70], reducing total dose and by reducing volume of irradiation. Dose reductions have been achieved for many tumors including retinoblastoma, pediatric soft tissue sarcomas of the head and neck, and certain CNS tumors including CNS germinoma. Volume reduction has been an important area of research in the treatment of medulloblastoma, ependymoma, low-grade astrocytoma, craniopharyngioma, and CNS germinoma [41, 45]. The risk of treating smaller volumes must be carefully balanced with objective gains documenting reductions in side effects in prospective clinical trials. To this end, the inclusion of endocrinology and its quantitative and relatively objective measures is essential. The risk of endocrine-related complications should be carefully considered in planning radiation therapy, but should not be used as a reason to avoid curative therapy. Careful follow-up and evaluation will lead to early intervention to mitigate consequences of irradiation.
5.3 Clinical Manifestations
5.3.1 GH Deficiency
Altered GH secretion leads to poor growth in childhood cancer survivors, particularly in young children after surgery in the suprasellar region, cranial irradiation [≥18 gray (Gy)], or total body irradiation (≥12 Gy). Hypothalamic function is affected more than is pituitary function. In most patients with GH deficiency, altered hypothalamic GHRH and somatostatin secretion lead to loss of the circadian pulsatile pattern of GH secretion. The radiation effect on GH secretion is dependent on fraction size and total hypothalamic dose-volume [46]. A large fraction size of radiation administered over a short period of time is more likely to cause GH deficiency than is the same total dose administered in smaller fractions over a longer period of time. The peak time for clinical identification of slowed growth consistent with GH deficiency is 3–5 years after such an insult, depending on RT dose. In one prospective study, all of the 21 children treated with a total dose of more than 45 Gy for optic pathway tumor experienced GH deficiency and significant slowing of growth rate within 2 years after irradiation [23]. At doses of cranial irradiation higher than 30 Gy (e.g., for suprasellar or posterior fossa tumor), the risk for GH deficiency may be more than 80 % by 10 years after RT [46]. Cranial irradiation doses greater than 24 Gy result in GH deficiency in as many as two thirds of patients who receive this treatment [13]. In many younger children, GH deficiency results from lower doses (>18 Gy). Doses of only 12–14 Gy of total body irradiation combined with chemotherapy and bone marrow transplantation also pose a significant risk for GH deficiency [13, 36, 37] (Table 5.3).
Growth rate is typically slow in children who are undergoing treatment for cancer and usually improves or shows catch-up after completion of cancer therapy (Fig. 5.8). Children whose growth rate does not improve or whose growth rate is less than the mean for age and gender should be evaluated for growth failure (Fig. 5.9). Causes of slow growth other than GH deficiency include hypothyroidism, radiation damage to growth centers of long bones or spine, chronic unresolved illness, poor nutrition, and depression. In individuals who have attained adult height, GH deficiency may be asymptomatic [24, 78], but alternatively may be associated with easy fatigability, decreased muscle with increased fat mass and truncal adiposity, and increased risk for cardiovascular disease [16, 25].
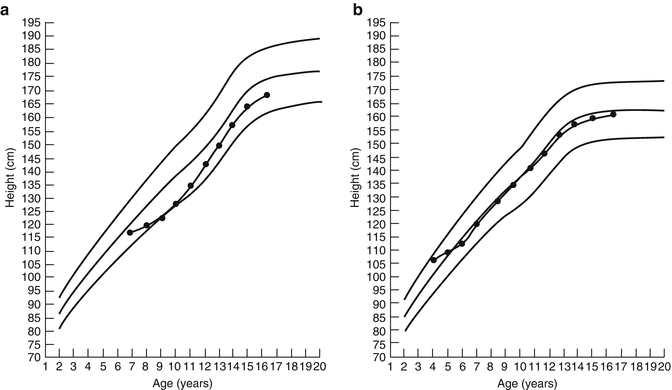
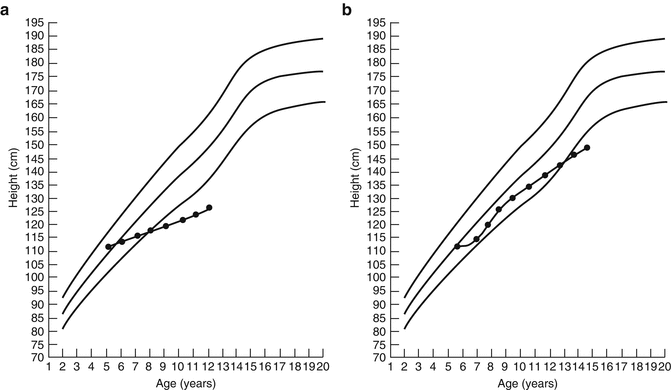
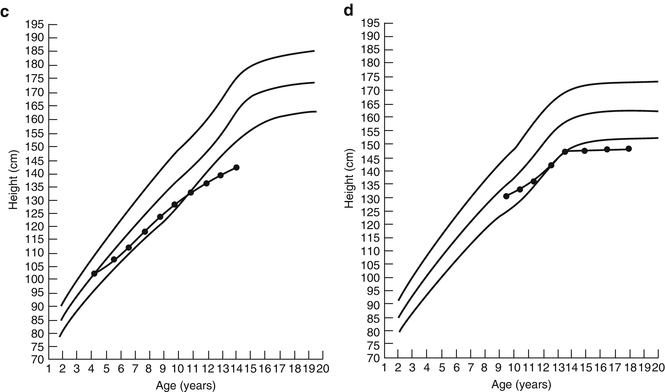
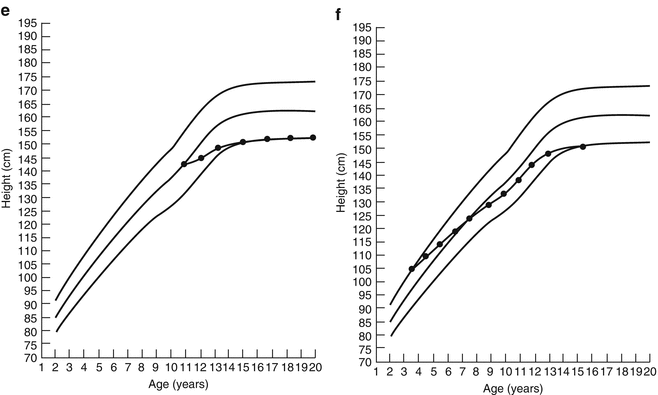
Fig. 5.8
(a) Complete catch-up growth in a boy after cancer therapy. (b) Growth in a girl after cancer therapy, without catch-up growth. Normal percentiles (5th, 50th, and 95th) as shown are obtained from the National Center for Chronic Disease Prevention and Health Promotion (2000)
Fig. 5.9
(a) Persistent growth failure in a boy after cancer therapy. (b) Later growth failure in a girl after recovery of normal growth. (c) Subtle persistent growth failure in a boy. (d) Growth in a girl with missed GH deficiency. (e) Growth in a boy with missed late onset GH deficiency. (f) Growth in a girl with central hypothyroidism
5.3.2 LH or FSH Deficiency
High doses of cranial radiation (≥30 Gy) are likely to cause hypothalamic GnRH deficiency and, therefore, gonadotropin deficiency (or in some patients, precocious onset of puberty through loss of inhibition that later progresses to gonadotropin deficiency through loss of GnRH secretory cells). Lower doses of cranial radiation (18–24 Gy) are more likely to cause damage to gamma-aminobutyric-acid-secreting neurons alone (leading to disinhibition and premature activation of GnRH neurons) and, therefore, rapid tempo of puberty or precocious puberty [3, 64]. In girls, the first signs of puberty are growth spurt and breast development (palpable breast buds or thelarche), followed by pubic hair growth and, after about 2 years, by menarche. In boys, the first sign of puberty is testicular enlargement (testes length >2.5 cm), followed by penile and pubic hair growth, followed by a growth spurt. In most studies of normal children, pubertal milestones are attained at ages that are normally distributed, with a standard deviation (SD) of approximately 1 year [76]. Children entering puberty more than 2 SDs earlier or later than average should be considered for endocrine evaluation. The average age that girls experience thelarche is 10 years and that of menarche is about 12 years; the average age when boys experience testicular growth is 11 years.
Patients with gonadotropin deficiency may have delayed, interrupted, or absent puberty. Staging of puberty is usually performed by the criteria of Tanner [76]. In survivors of childhood cancer, we initiate evaluation for delayed puberty in girls with no onset of breast development by 12 years of age or no menarche by 14 years of age and in boys with no sign of testicular growth by 13 years of age. Boys treated with agents that can cause infertility may have normal testosterone and LH concentrations, but reduced testicular volume and elevated FSH because of damage to the seminiferous tubules and reduced sperm production.
5.3.3 Precocious or Rapid Tempo of Puberty
Precocious puberty is defined as the onset of secondary sexual development before age 8 years in girls and before age 9 years in boys [9]. Despite controversy that puberty prior to these ages may occur in normal children [7, 69], younger occurrence of puberty than age 8 or 9 years may be the only clue to the presence of pathology and should not be ignored [48]. Pubic hair, acne, and body odor are not usually part of the presentation of precocious puberty in children younger than 4 years. Precocious puberty occurs in childhood cancer survivors who have lost inhibition of hypothalamic GnRH release as a result of tumor presence, raised intracranial pressure, cranial surgery, or low-dose cranial irradiation (18–24 Gy). Female gender and younger age at the time of cancer treatment are risk factors for precocious puberty. In some children who have received cranial irradiation, puberty may start at a normal age and advance rapidly. Thus, tempo of progression as well as timing of onset must be monitored. Rapid tempo of puberty is also caused by loss of inhibition of hypothalamic GnRH secretion. The outcome of early onset and/or rapid tempo of puberty can be short adult height and potential emotional lability; early bony maturation causes the child to lose the opportunity for 1–3 years of height growth (Fig. 5.10).
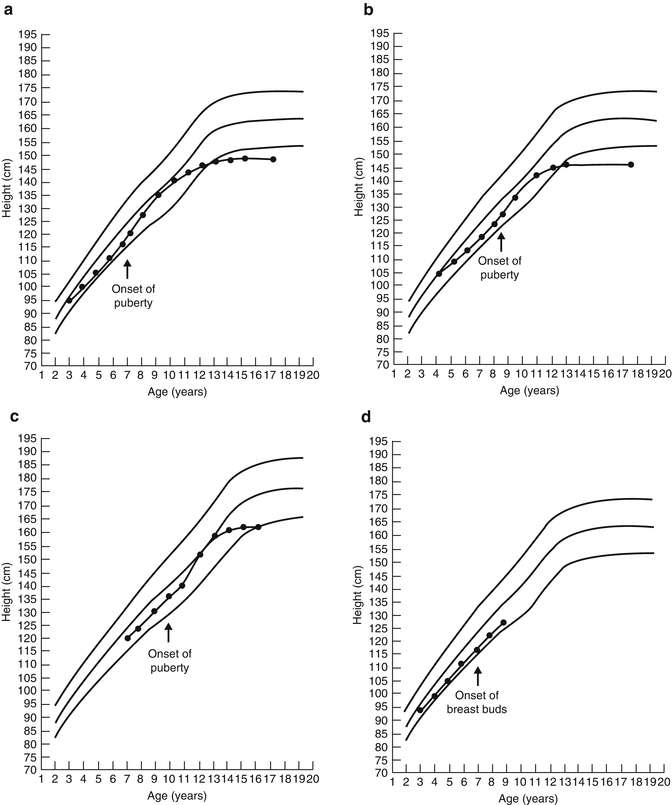
Fig. 5.10
(a) Growth in a girl with precocious puberty. (b) Growth in a girl with rapid/early puberty. (c) Growth in a boy with rapid/early puberty. (d) Growth in a girl with GH deficiency hidden by precocious puberty (no growth spurt)
5.3.4 Hypothyroidism
Central hypothyroidism refers to thyroid hormone deficiency caused by a disorder of the pituitary, hypothalamus, or hypothalamic-pituitary portal circulation. In contrast, primary hypothyroidism refers to under-function of the thyroid gland itself.
Primary hypothyroidism is the most common form of hypothyroidism in the general population and may occur in cancer survivors related both to family history and additional contribution from the cancer therapy. In primary hypothyroidism, the thyroid gland may have been injured through irradiation or autoimmune activity, but the central hypothalamic-pituitary-thyroid axis is intact.
In contrast, central hypothyroidism is characterized by blunted or absent nocturnal TSH surge, suggesting the loss of normal circadian variation in TRH release [58]. Central hypothyroidism is difficult to diagnose because of its subtle clinical and laboratory presentation. It is particularly difficult to recognize in patients whose growth is complete, because slowed growth rate can no longer be used as a sign. Symptoms of central hypothyroidism (e.g., asthenia, edema, drowsiness, adynamia, skin dryness) may have a gradual onset and go unrecognized until thyroid replacement therapy is initiated and the patient feels better [22]. In addition to causing delayed puberty and slow growth (Fig. 5.9f), hypothyroidism may cause fatigue, dry skin, constipation, increased sleep requirement, and cold intolerance. Central hypothyroidism was found in as many as 65 % of the survivors of brain or nasopharyngeal tumors, 35 % of bone marrow transplant recipients, and 10–15 % of leukemia survivors [56, 59].
Secretory dysregulation of TSH after irradiation may precede other endocrine disorders. In one cohort of patients with central hypothyroidism, 34 % had dysregulation of TSH secretion before the development of GH deficiency [58, 59].
In cancer survivors, mixed hypothyroidism reflects separate injuries to the thyroid gland and the hypothalamus (e.g., radiation injury to both structures). TSH values are elevated, but in addition, the secretory dynamics of TSH are abnormal with a blunted or absent TSH surge [58, 59]. This is in contrast to primary hypothyroidism in which TSH is elevated and the TSH surge is normal. In a study of 208 childhood cancer survivors referred for evaluation of possible hypothyroidism or hypopituitarism, mixed hypothyroidism was present in 15 (7 %) [59]. All of the patients with mixed hypothyroidism had free T4 concentrations in the low normal range.
5.3.5 ACTH Deficiency
ACTH deficiency is less common than other neuroendocrine deficits but should be suspected in patients who have a history of brain tumor (regardless of therapy modality), cranial irradiation, GH deficiency, or central hypothyroidism [63]. Though uncommon, ACTH deficiency can occur in patients who have received intracranial radiation that did not exceed 24 Gy, but occurs in less than 3 % of patients after chemotherapy alone [62, 63].
Symptoms of central adrenal insufficiency can be subtle and include poor weight gain, anorexia, easy fatigability, and poor stamina. In patients who have ACTH deficiency, as opposed to primary adrenal insufficiency, symptoms of salt craving, electrolyte imbalance, vitiligo, and hyperpigmentation usually are not observed. More overt manifestations of complete ACTH deficiency include weight loss and shakiness that is relieved by eating (hypoglycemia). Signs of adrenal crisis at times of medical stress include weakness, abdominal pain, hypotension, and shock.
Patients with partial ACTH deficiency may have only subtle symptoms unless they become ill. Illness can disrupt these patients’ usual homeostasis and cause a more severe, prolonged, or complicated course than expected. As in complete ACTH deficiency, incomplete or unrecognized ACTH deficiency can be life-threatening during concurrent illness. Death during sleep in hypopituitary patients has been proposed to be related to untreated ACTH deficiency [75].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


