Endocrine complications and paraneoplastic syndromes
Sai-Ching Jim Yeung, MD, PhD, FACP  Robert F. Gagel, MD
Robert F. Gagel, MD
Overview
Transformation of normal cells results in activation and/or suppression of a number of hormonally active genes. This chapter outlines the several clinical endocrine paraneoplastic syndromes and their appropriate management. Treatment of cancer results in a number of endocrine or metabolic manifestations, most of which are related to hormone deficiency or drug-related toxicity. The introduction of targeted therapy that disrupts signaling pathways and immunotherapy, contrary to expectations, has actually increased the number of endocrine manifestations. This chapter will chronicle the major new endocrine toxicities in addition to those observed with older cytotoxic chemotherapies and radiation.
Introduction
This chapter is divided into two major sections. The first focuses on endocrine complications in cancer patients and the second on endocrine paraneoplastic syndromes. Cancer and its treatment can lead to endocrine dysfunction or clinical and laboratory abnormalities that obscure or mimic endocrine diseases. Paraneoplastic syndromes are a group of diverse clinical syndromes seen in cancer patients caused by circulating biologic/humoral factors that include hormones, immunoglobulins, cytokines, and other agents.
Endocrine complications
Hypothalamic–pituitary dysfunction
Radiotherapy is a common cause of hypothalamic–pituitary dysfunction in cancer patients. There is no strong direct evidence to implicate chemotherapy as a cause of permanent dysfunction of the anterior pituitary, although some of the newer targeted therapies may affect pituitary function. Metastasis to the hypothalamic region or the pituitary gland is uncommon,1 and clinical manifestations of endocrine dysfunction because of metastatic disease in this region are rare. However, benign tumors such as pituitary tumors and craniopharyngiomas frequently affect this anatomic region and cause endocrine dysfunction. The introduction of immunotherapy by immune checkpoint blockade [anti-CTLA4 antibodies (ipilimumab and tremelimumab) and anti-PD-1 antibody (nivolumab)] has been associated with development of autoimmune hypophysitis that requires hormonal replacement (corticosteroids or thyroid hormone) in 2–3% of treated patients.
Development of radiation-induced hypothalamic dysfunction is insidious; hormonal deficiency can manifest years after radiation. In general, the rapidity of onset and severity of dysfunction depend on the total dose of radiation and the rate of delivery. The sequence and frequency of dysfunction among the axes of hypothalamic–pituitary functions vary. The somatotropic axis is the most susceptible, while the thyrotropic axis is the least susceptible (Figure 1).2–5 The diagnosis of hypothalamic–pituitary dysfunction requires vigilance of the physician because most presenting symptoms (e.g., fatigue and weakness) are nonspecific and attributable to other causes common among cancer patients. A diagnostic screen for hypothalamic/pituitary dysfunction may include serum growth hormone (GH) and insulin-like growth factor-1 (IGF-1) measurement and evaluation for gonadal failure. Signs of overt hypopituitarism include hypoglycemia, hypotension, and hypothermia.
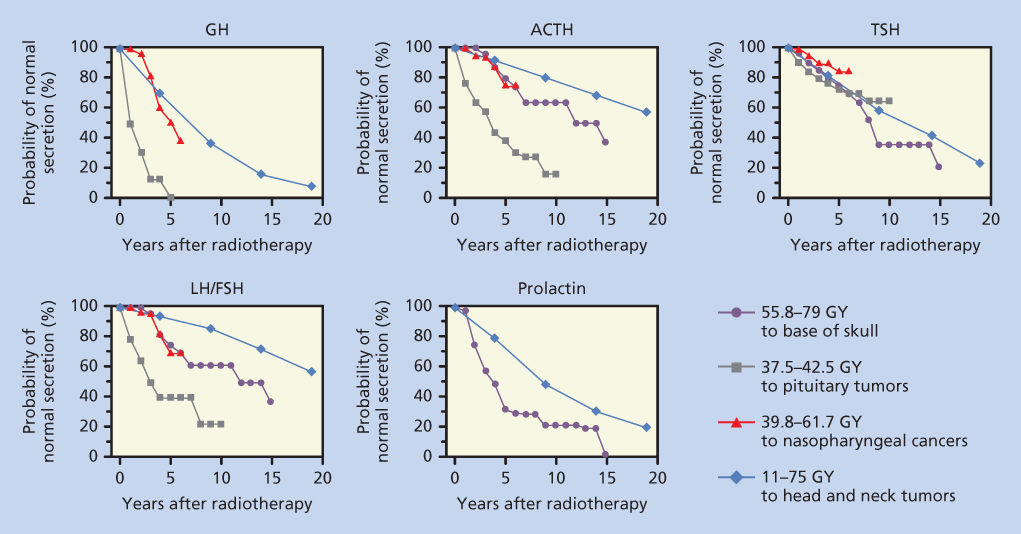
Figure 1 Probability of normal pituitary hormone secretion over time after radiation exposure to the hypothalamic–pituitary areas. Data from four studies were replotted on this single figure. The first set of values (closed circle) are from Pai et al.,3 where the patient received 55.8–79 Gy to the base of the skull. The second set of values (solid square) are from Shalet et al.,83 where patients with pituitary tumors were treated with 37.5–42.5 Gy. The third series (open triangle), from Lam et al.,2 shows the effect of radiation treatment for nasopharyngeal carcinoma with 39.8–61.7 Gy. The final series (open diamond) represents data from Samaan et al.,4 in which 11–75 Gy was administered to treat head and neck tumors.
In children and adolescents, evaluation of sexual development is a useful diagnostic tool. Staging sexual development according to Tanner’s criteria, menstrual history in girls and penile/testicular size in boys should be evaluated. In children who have had cranial irradiation, height and weight should be measured every 6 months. In children treated with spinal and craniospinal irradiation, local rather than general growth abnormalities may be present and, if so, require specific evaluation. Foot size is a reliable indicator of growth that can be easily measured. Deviation from normal growth curves should be evaluated for GH deficiency, hypothyroidism, and adrenal insufficiency. If the initial evaluation of GH, IGF-1, thyrotropin (TSH) and free thyroxine (T4) levels, and radiographic bone age reveal abnormality, then detailed dynamic testing to evaluate the hypothalamic/pituitary axes should be performed (Table 1).
Table 1 Dynamic testing of the hypothalamic/pituitary axes
| Test | Dose/sampling | Contraindications |
| Growth hormone axis | ||
| Insulin hypoglycemia | 0.075–0.1 U regular insulin/kg IV to achieve glucose ≤40 mg/dL. Sample for glucose and GH at 0, 30, 45, 60, and 90 min | Coronary heart disease or seizures |
| Arginine | 0.5 g/kg (up to 30 g) IV over 30 min. Sample for GH at 0, 30, 60, 90, and 120 min | Liver disease or renal disease |
| L-Dopa | 500 mg by mouth. Sample for GH at 0, 30, 60, 90, and 120 min | Systolic blood pressure <100 mm Hg or age > 60 years |
| Arginine and GHRH | Arginine dose as above. GHRH 1 µg/kg IV push. Sample for GH at 0, 30, 60, 90, and 120 min | Liver disease or renal disease |
| Clonidine stimulation protocol | Clonidine 0.15 mg/m2 by mouth. Collect GH samples at baseline, 30, 60, 90, and 120 min | |
| Growth hormone-releasing hormone (GHRH) stimulation protocol | GHRH at 1.0 µg/kg body weight IV push. Collect GH samples at baseline, 15, 30, 45, 60, 90, and 120 min | |
| Growth hormone suppression test | The test should be performed after an overnight fast with the patient maintained at bed rest. The patient should drink a solution of 100 g glucose. Collect GH samples at baseline, 60, and 120 min | |
| Adrenal axis | ||
| ACTH stimulation test, 1-h | Synthetic ACTH 1–24 1 or 250 µg IM or IV. Draw blood for cortisol at 30 and 60 min after injection | |
| ACTH stimulation test, 48-h | Beginning at 9 a.m., obtain baseline 24-h urine for 17-hydroxycorticosteroids (17-OHCS) and creatinine. Collect 24-h urine as on day 1. Beginning at 9 a.m., start IV and give 250 µg synthetic ACTH 1–24 in 250 mL normal saline over 8 h every 8 h for 48 h. Alternatively, 40 IU of depot formulation of purified bovine ACTH in gelatin IM every 12 h for 48 h. Repeat 24-h urine as on days 1 and 2 Days 4 and 5: Collect 24-h urine as on previous days | |
| Corticotropin-releasing hormone (CRH) stimulation test | Fast for at least 4 h before the test. Human CRH at 1.0 µg/kg IV bolus over 30 s. Blood samples should be collected at 15 and 1 min before CRH administration and at 15, 30, 45, 60, 90, and 120 min after for measurements of cortisol and ACTH | |
| Low-dose dexamethasone test, overnight | Dexamethasone 1.0 mg (adult) or 20 µg/kg (children) PO between 11 p.m. and midnight. Serum cortisol is collected at 8–9 a.m. the next morning. A cortisol level <1.8 µg/dL essentially excludes Cushing syndrome | |
| Low-dose dexamethasone test, 48-h | Serum cortisol is collected at 8–9 a.m. Dexamethasone 0.5 mg (adult) or 10 µg/kg (children) PO immediately after the cortisol is drawn and again every 6 h for 48 h. A second plasma cortisol is drawn at 9 a.m., 6 h after the last dexamethasone dose. Serum cortisol concentrations <1.8 µg/dL exclude Cushing syndrome | |
| High-dose dexamethasone test, 48-h | Serum cortisol is collected at 9 a.m. Dexamethasone is administered (2.0 mg; 50 µg/kg in children) every 6 h for 48 h. A second plasma cortisol is drawn at 9 a.m., 6 h after the last dexamethasone dose. Patients with functional adrenal adenomas show no suppression of cortisol levels in the 48-h sample relative to the initial (baseline) sample. Seventy-eight percent of patients with pituitary source of excess ACTH showed >50% suppression of plasma cortisol, while only 11% of patients with an ectopic source of excess ACTH had a >50% suppression | |
| Comprehensive, 6-day, low-/high-dose dexamethasone test | This protocol incorporates the low- and high-dose dexamethasone tests in succession. 24-h urinary free cortisol and/or 17-hydroxycorticosteroid (17-OHCS) measurement can help verify the results of serum cortisol and ACTH | |
| Gonadotropin-releasing hormone (GnRH) stimulation test | GnRH 100 µg IV. A sample for serum LH should be collected at baseline and 40 min after GnRH administration | |
| Metyrapone stimulation (overnight) test | At 11 p.m., metyrapone 30 mg/kg (maximum 3 g) PO with a snack. On the following morning, at 8 a.m., measure serum cortisol and 11-deoxycortisol | |
GH = growth hormone; GNRH = growth hormone-releasing hormone; IV = intravenous; IM = intramuscular; PO = by mouth.
In adults who have received cranial or head and neck irradiation, detection of hypothalamic–pituitary abnormalities is more challenging. One strategy to detect hypothalamic–pituitary abnormalities in adults consists of routine screening for GH deficiency and gonadal failure. It is recommended that measurements of IGF-1 and testosterone levels in males and documentation of menstrual history in females be obtained annually for 5 years, and then at 5-year intervals for another 10 years. Any abnormalities noted on the screening tests should be pursued with further dynamic testing to evaluate all the axes of hypothalamic–pituitary functions.
Immunotherapy-induced hypophysitis
The development of immunotherapeutic approaches to treat melanoma and a growing list of other malignancies has resulted in a number of bystander effects. Prominent among these side effects is immunotherapy-induced hypophysitis (IH). In the phase III clinical trial of ipilimumab, 2.3% of patients developed symptoms of IH and evidence of hormonal deficiency including glucocorticoid, thyroid, gonadal, and GH deficiency, necessitating long-term replacement therapy. The pituitary-related events were grade 3 or 4 in 1.9% of these patients, necessitating urgent treatment or hospitalization. The onset of pituitary failure ranged from 11 to 19 weeks, following initiation of ipilimumab therapy. Common presenting complaints include headache, mental status alterations, abdominal pain, modified bowel habits, and hypotension; it is often difficult to distinguish IH from brain metastasis or symptoms associated with widespread metastatic disease. This difficulty suggests that clinicians initiating therapy with immunotherapy agents should obtain baseline thyroid (free T4 and TSH), adrenal (ACTH, adrenocorticotropic hormone and 8 a.m. serum cortisol), gonadal [luteinizing hormone (LH), follicle-stimulating hormone, and estradiol in females or testosterone in males], and growth-related hormones (GH and IGF-1) as baseline before initiating therapy. These measurements should be repeated if a patient develops symptoms suggestive of endocrine deficiency or acute symptoms of hypophysitis (headache and visual disturbance). In patients with symptoms or physical findings consistent with IH, an MRI (magnetic resonance imaging) of the pituitary gland and repeat hormone testing as described above should be performed together with dynamic testing of the adrenal endocrine axis (e.g., low-dose cosyntropin stimulation test). A recent report highlighting a larger experience in a single center indicates that IH is observed not only with anti-CTLA4 agents (ipilimumab and tremelimumab),6 but also with an anti-PD1 agent (nivolumab). In a report describing 968 patients treated with these agents for melanoma, prostate cancer, and renal cell carcinoma at a single center, 2.7% (27 subjects) had one or two hormonal deficiencies together with either pituitary enlargement in MRI scans or headache.6 In these 27 patients, central adrenal deficiency was identified in 77%, central hypothyroidism in 89%, and central hypogonadism in 79%. An abnormality of the pituitary gland was found by MRI in 85%.6 After a median follow-up period of 17 months (range: 1–76), none of the patients recovered normal adrenocortical function, whereas 12–13% recovered thyroid or gonadal function. Clearly, this is a significant and evolving issue; it is unclear at this time whether combinatorial therapy with two or more immunotherapeutic agents, currently under development, will be associated with higher incidence and/or severity of toxicity.
Thyroid disorders
Thyroid disorders and abnormalities in thyroid function are commonly associated with cancer and its therapy.
Serum thyroid hormone-binding protein abnormalities
The levels of thyroid hormone-binding proteins [thyroxine-binding globulin (TBG), prealbumin, and albumin] can be modified by sex hormone levels and nutritional factors; abnormalities of both are encountered frequently in cancer patients. Several chemotherapy drugs affect thyroid function test results. L-Asparaginase appears to reversibly inhibit synthesis of albumin and TBG, resulting in low total thyroxine (T4), but normal free T4 levels.7 The combination of podophyllin and alkylating agents has also been reported to decrease TBG.8 Both 5-fluorouracil9 and mitotane10 increase the total T4 and triiodothyronine (T3) levels without suppressing TSH, suggesting that these drugs increase thyroid hormone-binding capacity in the serum.
Euthyroid sick syndrome
Alterations in thyroid hormone metabolism occur in patients with cancer and other serious systemic illnesses.11 Low serum T3 levels, which may be found in up to 70% of moderately to seriously ill cancer patients, are caused by a decrease in the extrathyroidal conversion of T4 to T3. Serum concentrations of free T4 are usually normal or high, while concentrations of free T3 are below normal or low. The patients are clinically euthyroid, and serum TSH level and TRH stimulation test results are normal.
In most patients with euthyroid sick syndrome, T3, T4, and TSH levels are normal. Clinical manifestations of hypothyroidism are usually absent, but assessment may be confounded by obtundation, edema, and hypothermia that may accompany severe illness. Low free T4 levels in the context of euthyroid sick syndrome usually indicate a grave prognosis, with a mortality rate of more than 50%. Although it is generally accepted that thyroid hormone therapy has no benefit, in practice it is sometimes difficult to differentiate between the euthyroid sick syndrome and secondary hypothyroidism. Judicious replacement of T4 at physiologic levels in these uncommon patients may be appropriate if there are no contraindications (e.g., active ischemic heart disease).
Hypothyroidism
Thyroidectomy
Thyroidectomy may be performed for a variety of oncologic reasons in the management of thyroid cancer, head and neck cancer, or thyroid metastasis. Thyroid replacement is needed in this group of patients. In thyroid cancer patients, supraphysiologic doses of thyroid hormone are adjusted to suppress TSH without overt hyperthyroid symptoms. In others, the dose of thyroid hormone should be adjusted to keep TSH in the normal range.
Radiation
Irradiation is an important cause of hypothyroidism [primary (thyroid), secondary (pituitary), and tertiary (hypothalamic)]. Radiation-induced primary hypothyroidism is caused by thyroid cell destruction, inhibition of cell division, vascular damage, and possibly an immune-mediated phenomenon. Factors that increase the risk of developing primary hypothyroidism include a high radiation dose to the vicinity of the thyroid gland, duration since therapy, lack of shielding of the thyroid during therapy, and combined irradiation and surgical treatments.12
The incidences of hypothyroidism after radiation therapy for various cancers and conditions are tabulated in Table 2.4, 12–22 A relationship between radiation dose and the prevalence of hypothyroidism is based on studies of patients with Hodgkin disease.15, 18 Long-term follow-up of patients treated with low-dose radiotherapy suggests that the threshold for causing clinically evident hypothyroidism is approximately 10 Gy. For Hodgkin-disease patients who received >30 Gy, the actuarial risk of hypothyroidism was up to 45%, 20 years after irradiation.15 Patients with frank or subclinical hypothyroidism should receive thyroid hormone-replacement therapy.
Table 2 Incidence of hypothyroidism (including compensated hypothyroidism) after radiotherapy.a
| Type of malignancy or conditions | Radiation dose | % with hypothyroidism |
| Hodgkin disease | 30–60 Gy | 30–50 |
| Head and neck cancer | 40–72 Gy | 25–50 |
| Lymphoma | 20–40 Gy (median 36 Gy) | 30–42 |
| Breast carcinoma | ? | 15–21 |
| Total-body irradiation in BMT | 13.75–15 Gy | 15–43 |
BMT = bone marrow transplantation.
a Data based on Refs 4 and 1213141516171819202122.
Chemotherapy
The diagnosis of hypothyroidism in 14% of BMT (bone marrow transplantation) patients who received chemotherapy but did not receive total-body irradiation23 suggests a causal relation between hypothyroidism and high-dose combination cytotoxic chemotherapy. This notion is also supported by studies that showed an increased incidence of primary hypothyroidism in patients treated with multiple combination drug regimens24, 25 with or without radiation.24 L-Asparaginase, in addition to inhibition of TBG synthesis discussed above, may also inhibit TSH synthesis reversibly and lead to temporary hypothyroidism with decreased free T4 levels.26
Thyroid dysfunction is a recognized side effect of cytokine treatments. Treatment with interleukin-2 produces thyroid dysfunction in approximately 20–35% of patients.27 These patients have hypothyroidism, hyperthyroidism, or hyperthyroidism followed by hypothyroidism.28 Approximately 10% of interferon-treated patients develop primary hypothyroidism.29 Pituitary enlargement secondary to interferon-induced hypothyroidism has also been reported.30 Patients with antithyroid antibodies before therapy are at higher risk of cytokine-induced thyroid dysfunction.
Retinoid X receptor (RXR) ligands may be used in the treatment of certain malignancies such as cutaneous T-cell lymphoma. Bexarotene (a RXR-selective ligand) caused secondary hypothyroidism dose-dependently.31 A single dose can rapidly suppress TSH in healthy subjects.32 In addition to suppressing transcription of TSH by an RXR-mediated thyroid hormone-independent mechanism, bexarotene also increases metabolic clearance of thyroid hormones by a nondeiodinase-mediated pathway.33
Tyrosine kinase inhibitors
The rapid introduction of small organic molecules that inhibit kinase activity (tyrosine kinase inhibitors, TKIs) into cancer therapy over the past decade has led to the recognition that many of these agents affect thyroid function in profound ways. The first mechanism is a direct effect of these agents on thyroid function. A number of TKIs target receptor tyrosine kinases with well-defined functions in the thyroid gland including the vascular endothelial growth factor receptor, the epidermal growth factor receptor, RET, KIT, and MET. Other agents target downstream signaling pathways such as RAF, PI3K, and mTOR. These signaling pathways are operative in the thyroid and important for normal thyroid cell growth and function. It is not surprising that disruption of these pathways has effects on thyroid cell growth and death and thyroid hormone synthesis. Indeed hypothyroidism occurs at significant incidence rates with a number of agents: sunitinib, 7–85%; pazopanib, 12%; nilotinib, 22%; axitinib, 20–100%; cabozantinib, 15%; sorafenib, 8–39%; dasatinib, 50%; and imatinib, 0–25%.34 Routine measurement of thyroid function (free T4 and TSH) at regular intervals during the first year of therapy with one of these agents seems prudent as it is often difficult to differentiate nonspecific side effects of TKI therapy from primary hypothyroidism or effects of metastatic cancer.
There is a second mechanism for development of hypothyroidism in patients treated with specific TKIs for metastatic thyroid carcinoma who have also undergone total thyroidectomy. Specific agents approved (vandetanib, cabozantinib, sorafenib, and lenvatinib) or studied (pazopanib, motesanib, and sunitinib) for treatment of differentiated or medullary thyroid carcinoma cause an elevation of the serum TSH concentration and low serum T4 levels with high percent incidences: vandetanib, 49%; cabozantinib, 57%; sorafenib, 41%; and lenvatinib, 57%. As each of these agents causes diarrhea and malabsorption, one presumed mechanism for this effect is reduced absorption of thyroid hormone, although effects on thyroid hormone metabolism have not been excluded. Increasing the thyroid hormone dosages by as much as one-third generally resolves the problem. It is important to note that there may also be malabsorption of calcium, magnesium, and vitamin D, causing worsening hypocalcemia or hypomagnesemia in patients with mild thyroidectomy-associated hypoparathyroidism.35 Routine measurement of free T4 and TSH should be incorporated into management as more than 50% of these patient will require a dose modification of thyroid hormone (as well as magnesium, calcium, and vitamin D supplementation). It is important to note that primary hypothyroidism, hypocalcemia, or hypomagnesemia can be readily treated, and their appearance, if addressed promptly, should not limit the use of the TKIs. However, in patients with hypothyroidism, hypocalcemia, or hypomagnesemia, all of which can prolong the QT interval, it may be necessary to withhold TKIs that prolong QT interval, that is, vandetanib,35 lenvatinib,36 and sorafenib in patients with QT prolongation until the QT abnormalities are resolved.
131I-containing compounds
The use of 131I for treatment of thyroid cancer requires a high serum TSH level. High TSH level is achieved by either withholding thyroid hormone replacement or increasingly by administration of recombinant human TSH. The use of 131I-containing compounds in the treatment of other tumors may result in hypothyroidism. For instance, using high-dose (100–1000 mCi) [131I]-metaiodobenzylguanidine to treat unresectable pheochromocytoma may result in primary hypothyroidism.37
Metastasis to the thyroid
Hypothyroidism secondary to metastatic infiltration and replacement of the thyroid by cancer is extremely rare.
Screening
Children who have received either head and neck or cranial irradiation should have a free T4 and a TSH measurement annually for 5 years, and every 2 years thereafter. Early detection of abnormal T4 and TSH levels will permit medical intervention before hypothyroidism adversely affects physical and intellectual development and growth. In adults, neck irradiation for treatment of lymphoma and various head and neck tumors is associated with a high incidence of primary hypothyroidism. Patients who have received irradiation should have free T4 and TSH levels measured annually for 5 years, and then every other year for 10 years, and thereafter every 5 years for another 10 years. Once hypothyroidism is diagnosed, the patient should receive thyroid hormone-replacement therapy.
Hyperthyroidism
Radiation-induced painless thyroiditis with hyperthyroxinemia is an uncommon side effect of external-beam radiotherapy to the head and neck area. Transient hyperthyroidism may occur as a result of inflammation and destruction of thyroid tissue with release of thyroglobulin (containing T4 and T3), and is usually followed by hypothyroidism. Transient hyperthyroidism has been reported after mantle radiotherapy in Hodgkin disease patients, and occurs usually within 18 months of treatment.38 A low uptake of radioiodine in most of these cases suggests a diagnosis of silent thyroiditis, but some have Graves’ disease. In one series of Hodgkin disease patient treated with radiation, the risk of Graves’ disease in these patients was estimated to be at least 7.2 times that in a healthy population.15
Ophthalmopathy similar to that in Graves’ disease has been reported within 18–84 months of high-dose radiotherapy to the neck for lymphoma, breast cancer, and nasopharyngeal or laryngeal cancer. Ophthalmopathy may occur without hyperthyroidism and in the absence of the human leukocyte antigen-B8.39 This suggests that radiation-induced thyroid injury may induce an autoimmune process that is similar to Graves’ disease.
There have also been examples of TKI-associated hyperthyroidism, including two deaths (sorafenib and sunitinib associated).40 The presumed mechanism of hyperthyroidism is thyroiditis. Patients with severe hyperthyroidism should be treated aggressively for thyroid storm; consideration should be given to holding TKI therapy until the patient is stabilized in such cases.34
Thyroid nodules and cancers
Low-dose radiation increases the risk of thyroid nodules and cancer. The association between thyroid cancer and low-dose irradiation has been extensively examined,41 and is discussed in the chapter about thyroid cancer (see 81).
Energy balance and glucose metabolism
Obesity and metabolic syndrome
Cancer treatments may lead to obesity and the metabolic syndrome.42–44 The metabolic syndrome is a cluster of abnormalities consisting of central obesity, dyslipidemia, hyperglycemia, and hypertension that increases the risk of type 2 diabetes and cardiovascular disease. Obesity is a modifiable risk factor for carcinogenesis as well as cancer progression. The mechanisms by which obesity promotes cancer include: hyperinsulinemia due to insulin resistance, high IGF-1, adipokines, low adiponectin, increased production of estrogens by adipose tissue, and increased inflammation. Obesity in cancer survivors may place them at increased risk for poor disease outcomes.42, 45, 46 Obesity increases the risk of colorectal and genitourinary second primary cancers.47 Although low levels of physical activity in cancer survivors can contribute to obesity, the pathophysiologic basis of the weight gain is unclear. As obesity is an adverse prognostic factor for many cancers and is a modifiable risk factor, secondary obesity after cancer treatment needs to be addressed.
Diabetes mellitus
Diabetes mellitus type 2 (DM2) is associated with an elevated risk of pancreatic, liver, colon, gastric, breast, and endometrial cancer.48–53 Extensive epidemiologic data suggest important roles of diabetes in carcinogenesis48–53 and cancer survival.54 The strongest association is perhaps with pancreatic cancer.55–59 Apart from the frequently coexisting obesity, the mechanisms by which diabetes promotes cancer include: hyperinsulinemia, high IGF-1, and hyperglycemia. Hyperglycemia per se has a promoting effect on cancer proliferation. In male, cancer survivors with a fasting serum glucose concentration ≥126 mg/dL had a higher relative risk for hepatopancreatobiliary second primary cancer.47 Evidence-based guidelines for the management of DM2 in cancer patients to optimize patient survival are lacking.
The administration of glucocorticoids (e.g., in combination therapy regimens, for edema of brain metastasis, for prevention of transplant rejection, for graft-versus-host disease in BMT, and for nausea/vomiting) is probably the most common cause of diabetes mellitus in cancer patients. Therefore, patients who receive glucocorticoids must be periodically screened for diabetes with evaluation of fasting glucose levels during therapy. Treatment with streptozocin60 or L-asparaginase61 may result in insulin-deficient diabetes mellitus. Although there is no evidence of a delayed onset of diabetes mellitus following treatment with streptozocin, follow-up has been limited and short term. For long-term survivors treated with streptozocin, periodic screening for delayed development of diabetes mellitus may be indicated. Diabetes mellitus may also develop as a consequence of serious pancreatitis secondary to treatment with L-asparaginase. Immunotherapy for cancer using cytokines such as interleukin-2 and interferons may cause toxicity to pancreatic β cells and lead to insulin-dependent diabetes.62 Tacrolimus, an immunosuppressive agent used to prevent graft-versus-host disease in BMT, also increases the incidence of diabetes, perhaps by damaging pancreatic β cells.63 Patients who received allogenic BMT are likely to be receiving both glucocorticoids, cyclosporine A and tacrolimus, and are particularly at risk for developing diabetes mellitus.64 Management of the blood glucose levels would depend on the severity of the blood glucose level abnormality and on the underlying pathophysiologic mechanism of the increase in blood sugar. In general, insulin will be needed in patients who are insulin deficient.
Metabolic bone diseases
Osteoporosis
Four groups of adult patients are at particular risk for accelerated bone loss and osteoporosis: (i) patients with lymphoma, myeloma, or leukemia; (ii) women with breast cancer treated with cytotoxic chemotherapy frequently undergo an early menopause65 and cannot receive estrogen-replacement therapy; (iii) postmenopausal women with estrogen-receptor positive breast cancer; and (iv) men with prostate cancer who are on antiandrogenic therapy and made hypogonadal. Normal bone remodeling involves a delicate balance between bone formation by osteoblasts and bone resorption by osteoclasts. Antineoplastic therapy is toxic to osteoblast function and decreases bone formation. Production by the tumor of hormonally active substances [e.g., parathyroid hormone-related protein (PTHrP), lymphotoxin, interleukin-1, and interleukin-6] may contribute to bone loss. In most cases, it is not clear whether bone loss is caused by antineoplastic therapy or by the underlying disease process and its effects (including cachexia, malnutrition, poor calcium and vitamin D intake, or a combination of these). In patients with breast or prostate cancer, sex steroid hormone deficiency induced by therapy is the most important cause of bone loss. Bone loss is prominent in patients with several disorders (myeloma, leukemia, and lymphoma), affecting the hematopoietic cells, perhaps because of cytokine production and an intimate relationship of hematopoietic cells with bone-forming cells or the use of high-dose or prolonged therapy with glucocorticoids.
A number of drugs can induce osteoporosis.66 In cancer patients, glucocorticoids, methotrexate, and cytotoxic drugs that cause renal loss of calcium, magnesium, or phosphorus (e.g., platinum compounds, cyclophosphamide, and ifosfamide) have significant impact on bone density. Osteoporosis (generalized or localized) is observed in children receiving methotrexate therapy for acute lymphoblastic leukemia (ALL).67 The osteoporosis improves significantly after cessation of methotrexate therapy. Longitudinal study of ALL patients showed that the leukemic process, high-dose glucocorticoids, and hypomagnesemia (owing to renal wastage following cyclical glucocorticoid and nephrotoxic chemotherapy or antiinfective agents) contributed to the impairment of calcium and vitamin D metabolism and decrease in bone mass at different stages of the treatment process.68 Adjuvant chemotherapy for breast cancer (usually involving 5-fluorouracil, cyclophosphamide, and doxorubicin or methotrexate) is associated with low bone mass in premenopausal patients.69 Bone loss during chemotherapy is substantial and may lead to increased risk of fracture. Posttreatment hypogonadism appears to be a major factor in these adult women with osteoporosis. While tamoxifen has a slight protective effect on bone loss, the opposite is true for aromatase inhibitors. BMT usually involves treatment with high-dose cytotoxic drug, glucocorticoids, and immunosuppressive agents. In 24 patients who underwent BMT with high-dose chemotherapy, profound effects on bone biomarkers were observed.70
Prompt investigation of gonadal dysfunction in cancer survivors and prompt replacement of gonadal steroids (in the absence of contraindications) in young hypogonadal men or women are recommended to decrease the risk of future bone fractures. The bone mass of long-term cancer survivors should be assessed when the patient is about 30 years old, the age at which most people have attained peak bone mass.71 If bone mass is normal, no further evaluation is needed beyond the usual recommendations for prevention of osteoporosis. If it is abnormal (more than 2 standard deviations below normal), the patient should be referred for evaluation of the multiple reversible causes of osteoporosis.
A key point in the management of the osteoporosis syndrome in cancer patients is the use of bone mineral density measurement (e.g., by dual-energy X-ray absorptiometry) to assess fracture risk and to monitor the effects of therapy. This measurement should be performed early in the course of management of the malignancy so that appropriate preventive measures can be implemented. The oncologist who is prescribing medications that are likely to decrease bone mass should consider active use of bisphosphonates (e.g., alendronate, risedronate, ibandronate, or zoledronate), calcitonin, selective estrogen-receptor modulators (SERMs), or denosumab, in addition to a daily intake of 1200–1500 mg elemental calcium and vitamin D supplementation. Bisphosphonates and denosumab are effective therapies for prevention of bone loss. While osteoporosis in children with leukemia will frequently reverse because the children are in the formative years of bone development, in adults more active measures such as bisphosphonate or teriparatide therapy to prevent bone loss should be considered, rather than waiting for the development of a fracture syndrome.
Another key point is correction of abnormal mineral and vitamin D metabolism by dietary supplements. Nutritional deficiency in teenagers and young adults results in lower bone mass. Treatment of hypocalcemia, hypomagnesemia, and vitamin D deficiency is integral to the successful therapy of osteoporosis in cancer patients. Recent studies document that clinically relevant vitamin D deficiency is present in 50% of the normal population; the percentage is almost certainly higher in patients undergoing cancer therapy.
Osteomalacia
Osteomalacia, a condition characterized by unmineralized bone matrix, is a rare complication of chemotherapy, but should be considered in osteopenic patients and those with osteomalacic clinical syndrome (bone pain and proximal myopathy). The most common cause is a decrease in the serum calcium and/or phosphorus concentrations caused by nutritional deficiency and renal wasting of phosphorus and calcium. Patients who have received chemotherapeutic agents that cause hypophosphatemia, hypomagnesemia, or hypocalcemia are particularly at risk. Investigation of the levels of serum ionized calcium, phosphorus, magnesium, and vitamin D metabolites should be included in the initial evaluation. Appropriate replacement therapy of these vitamins and minerals should be instituted once deficiencies have been identified. Other contributing factors include systemic acidosis and drugs such as anticonvulsants and aluminum.66 Tumor-induced osteomalacia will be addressed in the section that follows discussing paraneoplastic syndromes.
Ifosfamide causes tubular damage leading to renal phosphate wasting, hypophosphatemia, and rickets/osteomalacia.72 The toxic effects of ifosfamide on renal tubular function include Fanconi syndrome in adults and children. Tubular damage is seen most commonly when ifosfamide is administered in doses of 50 g/m2 or more, or when it is used in combination with cisplatin.73 Rickets is reported most commonly in children. Estramustine, used in the treatment of prostate cancer, has been reported to increase bone resorption and at the same time cause hypocalcemia, hypophosphatemia, and secondary hyperparathyroidism.74
Adrenal diseases
Adrenal metastasis
Hematogenous metastasis to the adrenal glands is common, exceeded in frequency only by hematogenous metastasis to the lung, liver, and bone.75 Autopsies have documented that 9–27% of patients who died from malignant illness had adrenal metastasis, with bilateral involvement in one-half to two-thirds of patients with adrenal metastasis.
The presence of adrenal metastasis may have important implications for diagnostic and therapeutic planning. When patients with cancer have an adrenal mass but no evidence of metastasis elsewhere, it is important to determine whether this mass represents a metastatic tumor or a separate, unrelated adrenal lesion. Recent advances in imaging techniques have allowed the identification of adrenal lesions antemortem as part of the tumor-staging evaluation. The location of the adrenal glands in the perinephric fat allows the detection of almost all normal glands and contour-deforming masses as small as 5–10 mm. Computed tomography (CT) has a sensitivity and specificity in the detection of adrenal masses. Characteristics on CT examination that suggest adrenal metastasis rather than primary adrenal disease include heterogeneity, contrast enhancement, bilaterality, and size >3 cm.76
Without other evidence of metastatic disease, whether the adrenal mass is actually a metastatic tumor is critical information in determining the appropriate therapy for the cancer. Evaluation of a patient who has a malignant adrenal mass should include a history and physical examination to elicit evidence of adrenal insufficiency, Cushing syndrome, mineralocorticoid excess, or pheochromocytoma. Biochemical assessment should include a short ACTH stimulation test with measurements of serum cortisol to rule out adrenal insufficiency. A 24-h urine collection should be obtained to measure urinary free cortisol, aldosterone, catecholamines, and metanephrines. Pheochromocytoma must be excluded, especially if there is hypertension, or an operative procedure of any type is contemplated. It has been reported that one-half of the patients who had a clinically unsuspected pheochromocytoma had clinical deterioration or even death immediately following a non-adrenal-related surgical procedure.77
If the biochemical assessment for pheochromocytoma is negative, CT-guided fine-needle aspiration should be considered. This procedure has a sensitivity of 85% in detecting cancer.78
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


