Chapter 10 Emtricitabine
Emtricitabine is a pyrimidine analog with antiviral activity against human immunodeficiency virus (HIV) and hepatitis-B virus (HBV) whose activity was originally described by Schinazi et al in 1992.1 Emtricitabine received approval from the US Food and Drug Administration (FDA) for use as a therapeutic agent for the treatment of HIV on 2 Jul 2003. Guidelines for the treatment of HIV infection by both the US Department of Health and Human Services (DHHS)2 and the International AIDS Society-USA (IAS-USA)3 have placed emtricitabine in the ‘preferred category’ of nucleoside reverse transcriptase inhibitors (NRTIs) in combination with tenofovir, zidovudine, or didanosine (IAS-USA guidelines only) for antiretroviral-naive patients initiating combination antiretroviral therapy. While emtricitabine has not received FDA approval for the treatment of HBV-infections or HIV–HBV co-infections, it is often used ‘off label’ in this capacity.
STRUCTURE
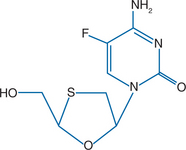
Emtricitabine (FTC), the negative or cis enatiomer of 2′,3′-dideoxy-5-fluoro-3′-thiacytidine, has antiviral activity against HIV-1, HIV-2, and HBV.1,4 Similar to lamivudine, emtricitabine is an oxathiolane-cytosine analog and has the unusual L configuration (1-β-L) rather than the D configuration found in natural nucleosides. Unlike lamivudine, emtricitabine has a fluorine in the 5-position of its cytosine ring (Fig. 10-1).
MECHANISM OF ACTION AND IN VITRO ACTIVITY
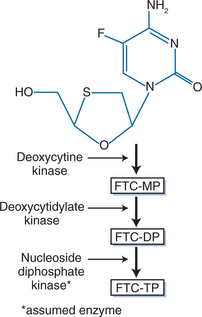
Like other nucleoside analogs, emtricitabine is a prodrug that must be metabolized intracellularly to exert its activity. After entering the cell through multiple transport mechanisms, emtricitabine is phosphorylated stepwise by cellular kinases to its active 5′-triphosphate form, emtricitabine 5′-triphosphate (E-TP) (Fig. 10-2).5–7 E-TP competitively inhibits the binding of deoxycytidine triphosphate, an endogenous deoxynucleotide triphosphate, to the reverse transcriptase (RT) binding site.8,9 E-TP is incorporated by RT into the nascent proviral DNA chain. This incorporation causes termination of DNA synthesis because E-TP lacks a hydroxyl group in the 3′ position of its sugar moiety and therefore does not allow the 5′ to 3′ linkage required for DNA synthesis. Thus, incorporation of E-TP inhibits viral replication.8–10
Emtricitabine’s antiviral activity has been evaluated in lymphocyte and monocyte/macrophage cell lines and peripheral blood mononuclear cell (PBMC) specimens. Emtricitabine is active against laboratory-adapted strains of HIV-1 and HIV-2 as well as clinical isolates of HIV-1, including zidovudine- and didanosine-resistant isolates. Depending on the cell type and viral isolate used, the concentration required to produce 50% inhibition (IC50) of HIV ranges from 0.0014 to 1.500 μM.1,11–15 Emtricitabine’s generally low IC50 values are attributed to its ability to bind efficiently to the RT-DNA complex. In a kinetic analysis of RT-catalyzed DNA synthesis, E-TP bound 3.6-fold tighter to the enzyme–DNA complex (Kd, rate of dissociation) and was incorporated into nascent chain DNA (Kpol, rate of polymerization) 2.5-fold faster than was lamivudine 5′-triphosphate (L-TP) (Table 10-1).16

In addition to HIV-1 and HIV-2, emtricitabine also inhibits HBV in vitro. Its in vitro anti-HBV activity is comparable to that seen with lamivudine, inhibiting virus production with an IC50 of 0.01-0.04 μM. Intracellularly, emtricitabine inhibits HBV DNA synthesis with an IC50 of 0.16 μM.6,17,18
When inhibiting HIV-1, emtricitabine is synergistic in vitro with the nucleoside analogs zidovudine14 and stavudine.19 Using MT-2 cells infected with HIV-1 strain IIIB (a zidovudine-sensitive isolate), synergistic antiviral activity occurred when emtricitabine was combined with zidovudine or stavudine. Additive antiviral activity occurred when emtricitabine was combined with zalcitabine or didanosine.19
PHARMACOKINETICS
The pharmacokinetic (PK) properties of emtricitabine have been well characterized.20 Emtricitabine is rapidly and well absorbed following oral doses with an oral bioavailability of greater than 90%. When single doses of emtricitabine were given orally to HIV-infected volunteers over the dose range of 100–1200 mg, emtricitabine disposition followed linear kinetics, and the steady-state emitricitabine concentrations were predictable based on single-dose data. Plasma emitricitabine concentrations reached levels above the in vitro IC50 and IC90 values even after single-dose administration. Emtricitabine’s bioavailability was not significantly affected by food intake.
Based on studies in healthy or HIV-infected adults administered emtricitabine 200 mg once daily as monotherapy or in combination with other antiretrovirals, the mean steady-state peak plasma concentration of ∼2 mg/mL occurs within 2 h after the dose. The mean steady-state peak plasma area under the curve (AUC) over the 24-h dosing interval is ∼7–11 h μg−1 mL−1 and mean half-life (t½) is ∼7.5–10 h.20,21 Comparable PK characteristics are seen in neonates less than 3 months of age given 3 mg/kg qd,22 and in children administered a dose of 6 mg/kg qd.23
Emtricitabine is excreted predominantly in the urine. Following a single oral dose, 86% of the emtricitabine dose was recovered in the urine and 13% in the feces. More than 85% of the dose was excreted in the urine as unchanged emitricitabine. The effect of renal impairment and hemodialysis on emtricitabine’s PK parameters has been studied, and dose adjustments are recommended for patients with baseline creatinine clearance <50 mL/min including patients on hemodialysis.24
E-TP has a prolonged intracellular half-life compared to its plasma half-life and the intracellular half-life of L-TP. The estimated intracellular half-life of E-TP is 39 h,20 compared with 16 h for L-TP (Table 10-1).25
Among women, emtricitabine achieves higher levels in genital tract secretions relative to plasma levels following both a single dose and at steady state (6.1-fold and 6.7-fold higher AUC ratios, respectively).26 This may have implications for its use in the prevention of mother-to-child transmission and in postexposure prophylaxis regimens.
During drug development, emtricitabine was hypothesized to penetrate the central nervous system because of the likely lipophilicity of its fluorine;1 however, studies have shown that emtricitabine poorly penetrates into the cerebrospinal fluid (CSF). Animal studies have shown that emtricitabine does not effectively cross the blood–brain barrier. In monkeys, the concentration of emtricitabine in the CSF was only 4.0 ± 0.7% of the corresponding plasma emtricitabine level.27
TOXICITY
Emtricitabine, like other nucleoside analogs, has the potential for toxicity due to interaction with human DNA polymerases. Of particular concern is the potential for interactions with human mitochondrial DNA complexes, such as mitochondrial DNA polymerase-γ, as disruption of mitochondrial enzymes by NRTIs has been implicated as a cause of hepatic steatosis, primary lactic acidosis syndrome, myopathy, and neuropathy.28–30 In light of this concern and because of the significant hepatotoxicity observed in subjects treated with another fluorinated nucleoside, fialuridine (FIAU),31 emphasis has been placed on investigating emtricitabine’s effects on cellular and mitochondrial DNA polymerases in vitro and on emtricitabine’s toxicities in clinical trials.
In Vitro Studies
In investigations performed in vitro, E-TP does not appear to interfere significantly with cellular and mitochondrial DNA polymerase function. When the antiviral activity of E-TP against RT was compared with E-TP’s activity against human (HeLa cell) DNA polymerases α, β, γ, and ε, E-TP was a weak inhibitor of each of these enzymes when compared to its inhibition of HIV RT. Apparent Ki values were 6.0 μM for polymerase-α, 17 μM for polymerase-β, 6.0 μM for polymerase-γ, and 150 μM for polymerase-ε, compared to the Ki value of 0.17 μM for HIV-1 RT.32 When the activity of E-TP against mitochondrial DNA polymerases (pol γ) was compared with lamivudine’s effect, E-TP was found to incorporate into mitochondrial DNA at a rate that was 15-fold slower than that of lamivudine (Kpol), while the affinity of E-TP for the enzyme–DNA complex was sevenfold weaker (Kd) (Table 10-1).33
Animal Studies
In short- and long-term animal toxicology studies in mice, rats, and cynomolgus monkeys, emtricitabine showed little toxicity. The only adverse outcome was mild reversible anemia.32,34 In mice and rabbits exposed to emtricitabine in concentrations 60-fold (mice) and 120-fold (rabbits) higher than human exposures at the recommended daily dose (200 mg), emtricitabine did not adversely affect fertility or cause teratogenesis. It did not adversely affect postnatal development, neurologic development, or learning in F1 mice.35
Human Studies
In humans, emtricitabine does not significantly affect general behavior or neurologic, autonomic, central nervous system, cardiovascular, renal, gastrointestinal, or smooth muscle function. Selected adverse events occurring with at least a 5% frequency in controlled studies of emtricitabine include abdominal pain (8–13%), asthenia (10–16%), headache (12–22%), fever (2–8%), diarrhea (9–24%), nausea (14–20%), vomiting (8–12%), arthralgia (3–6%), depression and/or anxiety (1–7%), insomnia (0–7%), cough (4–14%), rhinitis (4–19%) and pharyngitis (4–13%).36–40 Few serious adverse events with emtricitabine have been reported to date. Selected grade 3/4 laboratory abnormalities occurring with at least a 5% frequency in controlled studies of emtricitabine include elevated creatine kinase (11–12%), AST (3–6%), ALT (2–5%), triglycerides (9–10%), and neutropenia (5%).36–40 In studies comparing emtricitabine and lamivudine, adverse events and grade 3/4 laboratory abnormalities, including those requiring discontinuation of study medication, are comparable and infrequent.37 In pediatric studies, no adverse events or laboratory abnormalities occurs with a frequency greater than 5%. In children, the most common laboratory abnormalities were leucopenia (3%) and ALT elevation (3%).41
Skin discoloration is a unique toxicity of emtricitabine therapy that has been observed in ∼3% of subjects receiving the drug in clinical trials.42 Racial/ethnic differences in the frequency of this adverse event have been observed with skin discoloration occurring in 8% of blacks, 0.2% of whites, 2.7% of Hispanics, and 3.8% of Asians. This adverse event typically manifests as hyperpigmentation on the palms and soles, is usually of mild degree, and has not been associated with long-term toxicities to date. Discoloration of the tongue, arm, lip, and nail beds have also been observed, although this is less common (<1% frequency). The hyperpigmentation/discoloration resolves in some patients who discontinue treatment, and in some patients who continue to receive emtricitabine.43
Emtricitabine is a pregnancy category-B drug, and as such is acceptable to use during pregnancy.24 Over 70 women have become pregnant during clinical trials of emtricitabine. None of the infants born to exposed mothers had congenital abnormalities.43
ANTIRETROVIRAL ACTIVITY AND CLINICAL EFFICACY
Emtricitabine Monotherapy
Adult volunteers, adult HIV-infected patients, and HIV-infected or HIV-exposed pediatric patients have received emtricitabine in phase I/II clinical trials. Most of these trials are PK and dose-finding trials. However, studies FTC-10144 and FTC-10245 also evaluated the antiviral activity of emtricitabine alone and in comparison with lamivudine. FTC-101 was a phase I/II, open-label, dose-range study of 41 treatment-naive, HIV-infected subjects who were administered emtricitabine monotherapy at 25 mg bid, 100 mg qd, 100 mg bid, 200 mg qd, and 200 mg bid doses for 14 days.44 Antiretroviral suppression (1.3 log10) occurred in all dosage cohorts, with a strong trend toward greater activity at the high doses (200 mg bid and 200 mg qd). FTC-102 was a randomized, open-label study of 81 HIV-infected patients given 10 days of monotherapy with emtricitabine or lamivudine.45 The antiretroviral activity and safety of emtricitabine at doses of 50 mg qd, 100 mg qd, and 200 mg qd were compared with lamivudine 150 mg bid. Subjects receiving emtricitabine 200 mg qd had a significantly greater change in plasma HIV RNA, as measured by the average AUC minus baseline plasma HIV RNA level (AAUCMB) through day 11, compared with subjects treated with lamivudine (P = 0.04). The mean reduction in plasma HIV RNA levels was 1.7 log10 for subjects receiving emtricitabine, compared with 1.5 log10 for subjects receiving lamivudine (Table 10-1). These studies confirmed the potency of emtricitabine when used as a once-daily drug.
Emtricitabine in Three-Drug Combinations
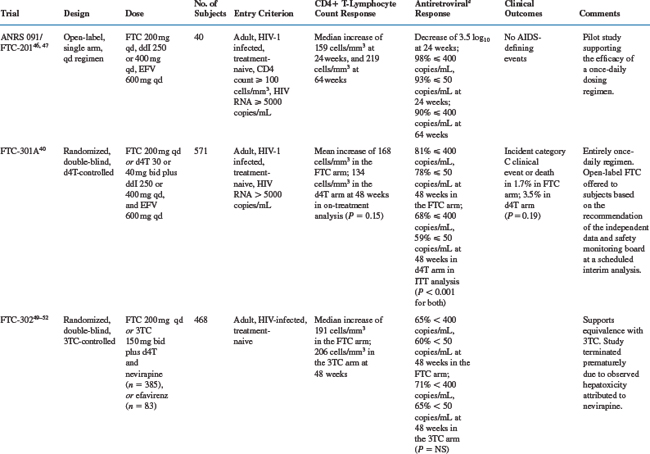
As shown in Table 10-2, several studies have evaluated the efficacy and safety of emtricitabine in combination with other antiretrovirals.


ANRS 091/FTC-201 Study
The ANRS 091/FTC-201 study, a phase II pilot study, assessed the activity of a once-daily antiretroviral regimen that combined emtricitabine, didanosine, and efavirenz in treatment-naive subjects.46 Among the 40 study subjects, the median baseline CD4+ T-lymphocyte count was 373 cells/mm3, and the median baseline plasma HIV RNA was 4.8 log10 copies/mL. After 24 weeks of treatment, the median increase in CD4+ T-lymphocyte count was 159 cells/mm3, and the median decrease in plasma HIV RNA was 3.4 log10. At 24 weeks, 98% of subjects had plasma HIV RNA levels of less than 400 copies/mL, and 93% had levels of less than 50 copies/mL. At 64 weeks the median increase in CD4+ T-lymphocyte count was 219 cells/mm3, and 90% of subjects had plasma HIV RNA levels of less than 400 copies/mL.47 No significant toxicities were reported during this trial.
FTC-301A Study
In the FTC-301A study, a total of 580 treatment-naive subjects were randomized to FTC or stavudine in combination with didanosine and efavirenz in a placebo-controlled study.40 Among the 571 subjects who received study drugs and were included in analyses, mean baseline log10 plasma HIV RNA levels were 4.8 copies/mL and mean baseline CD4+ T-lymphocyte counts were 312 cells/mm3 and 324 cells/mm3 in the emtricitabine and stavudine arms respectively. Eighty-one percent of FTC treated patients and 68% of stavudine treated patients completed 48 weeks of randomized, blinded therapy. Seven percent of emtricitabine-treated patients and 13% of stavudine-treated patients discontinued study therapy due to adverse events. Four percent of patients in the emtricitabine arm and 11% of patients in the stavudine arm discontinued treatment due to virologic failure. Following a planned interim analysis by an independent data and safety monitoring board when all patients had completed 24 weeks of therapy, the double-blind comparative phase of the study was terminated and all patients in the group demonstrating lesser response were offered open-label emtricitabine. Blinding of the original treatment assignment was maintained until all patients completed their week 48 visit. Only four subjects opted to receive open-label emtricitabine, and all four patients did so on or after their week 44 study visit. After 48 weeks on study medications, the percentage of patients with plasma HIV RNA <400 copies/mL were 81% in the emtricitabine arm and 68% in the stavudine arm (P < 0.001), and with plasma HIV RNA <50 copies/mL were 78% and 59%, respectively (P < 0.001). The mean increase in CD4+ T-lymphocyte count was not significantly different between the treatment arms in on-treatment analysis (168 cells/mm3 in the emtricitabine group and 134 cells/mm3 in the stavudine group, P = 0.15). In intention-to-treat sensitivity analysis with the last CD4 count carried forward to week 48 for those subjects withdrawing from the study, a statistically significant difference in mean increase in CD4+ T-lymphocyte count between the treatment arms was observed (153 cells/mm3 in the emtricitabine group and 120 cells/mm3 in the stavudine group, P = 0.02). No difference in clinical progression, defined as development of a new clinical event included in category C of the 1993 classification of the US Centers for Disease Control and Prevention (CDC),48 or death due to any cause, between the treatment arms was observed (1.7% in the emtricitabine group and 3.5% in the stavudine group, P = 0.19).
FTC-302 and FTC-303 Studies
Two randomized, equivalence (noninferiority) studies have compared the relative antiretroviral activity of emtricitabine and lamivudine. The FTC-302 trial, a double-blind, placebo-controlled study, randomized a total of 468 treatment-naive subjects to emtricitabine- or lamivudine-containing regimens.49,50 At baseline, the mean log10 plasma HIV RNA levels were 4.5, and the mean CD4+ T-lymphocyte counts were 392 cells/mm3 and 386 cells/mm3 in the emtricitabine and lamivudine arms. During the study, 10% of subjects receiving emtricitabine and 10% receiving lamivudine experienced adverse events and withdrew from study; 12% of both groups withdrew for other reasons. After 48 weeks on study medications, the percentages of study subjects with plasma HIV RNA levels of less than 400 copies/mL and less than 50 copies/mL did not significantly differ between the two groups, (emtricitabine: 65% <400 copies/mL and 60% <50 copies/mL; lamivudine: 71% <400 copies/mL and 65% <50 copies/mL).49 Virologic failure, defined as a lack of response (plasma HIV RNA >400 copies/mL at week 12) or loss of response (two consecutive plasma HIV RNA levels >400 copies/mL after having reached <400 copies/mL), occurred in 14% of the emtricitabine group and 10% of the lamivudine group.51 Of note, FTC-302 was terminated prematurely due to hepatotoxicity attributed to nevirapine.52
The FTC-303 study randomized 440 subjects on lamivudine-containing regimens with plasma HIV RNA levels of less than 400 copies/mL for at least 12 weeks to open-label emtricitabine or continued lamivudine in a 2:1 allocation.38 At baseline, subjects had received lamivudine-containing regimens for a median of 35 months; 79% were on protease inhibitor (PI)-containing regimens, and 21% were on non-nucleoside reverse transcriptase inhibitor (NNRTI)-containing regimens. Baseline plasma HIV RNA levels were less than 50 copies/mL in 86% and 50–400 copies/mL in 14%. A significantly higher percentage of subjects receiving emtricitabine withdrew owing to adverse events compared to those receiving lamivudine (4% vs 0%). After 48 weeks on study medications, the percentages of study subjects with plasma HIV RNA levels of <400 copies/mL and <50 copies/mL were not significantly different between the two groups (emtricitabine: 77% <400 copies/mL and 67% <50 copies/mL; lamivudine: 82% <400 copies/mL and 72% <50 copies/mL). Virologic failure, defined as plasma HIV RNA levels of more than 400 copies/mL on two consecutive mea-surements, occurred in 7% of subjects receiving emtricitabine and 8% of subjects receiving lamivudine.




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


