James S. McCarthy, Glenn W. Wortmann, Louis V. Kirchhoff
Drugs for Protozoal Infections Other Than Malaria
This chapter includes the antiprotozoal drugs other than those used to treat malaria, namely drugs to treat infections caused by Leishmania parasites, African and American trypanosomes (causes of sleeping sickness and Chagas’ disease, respectively), and drugs used to treat intestinal protozoal infections, particularly giardiasis and amebiasis. The drugs of choice for these infections are summarized in Table 41-1. Doses and alternative drugs are given in this chapter as well as in the chapters describing the parasites. Antiprotozoal drugs with antimalarial activity are covered in Chapter 40, sulfonamide drugs are covered in Chapter 33, and metronidazole is covered in Chapter 28.
TABLE 41-1
Drugs for Protozoa Other Than Malaria
| INFECTING ORGANISM | TREATMENT OF CHOICE | ALTERNATIVE TREATMENTS |
| Leishmaniasis: Leishmania donovani, L. major, L. infantum, L. chagasi, L. mexicana, L. tropica, L. (Viannia) spp. | ||
| Cutaneous leishmaniasis | Sodium stibogluconate or meglumine antimoniate, 20 mg/kg/day IV or IM × 20 days, or liposomal amphotericin B, 3 mg/kg/day IV × 7-10 doses* | Miltefosine, 2.5 mg/kg/day (max. 150 mg) PO × 28 days |
| Mucosal leishmaniasis | Liposomal amphotericin B at 20-35 mg/kg (total dose) given as 3 mg/kg IV once daily | Sodium stibogluconate or meglulmine antimoniate 20 mg/kg/day IV or IM × 28 days; miltefosine 2.5 mg/kg/day (max. 150 mg) PO × 28 days; amphotericin B deoxycholate, 1 mg/kg IV qod or daily to a total dose of 20-40 mg/kg |
| Visceral leishmaniasis | Liposomal amphotericin B, 3 mg/kg/day IV × days 1-5, then at day 14 and 21 | Sodium stibogluconate or meglumine antimoniate, 20 mg/kg/day IV or IM × 28 days; miltefosine 2.5 mg/kg/day (max. 150 mg/day) PO × 28 days; amphotericin B deoxycholate, 0.5-1 mg/kg/day or qod to a total dose of 15-20 mg/kg* |
| Amebiasis (Entamoeba histolytica) | Tinidazole, 2 g PO once daily × 3-5 days | Metronidazole, 500-750 mg PO or IV tid × 7-10 days |
| Giardiasis (Giardia lamblia, also called G. duodenalis or G. intestinalis) | Tinidazole, 2 g PO once | Metronidazole, 250 mg PO tid × 5-7 days |
| Trichomoniasis (Trichomonas vaginalis) | Tinidazole, 2 g PO once | Metronidazole, 2 g PO once |
| Cryptosporidiosis (Cryptosporidium spp.) | Nitazoxanide, 500 PO bid × 3 days† | |
| American trypanosomiasis (Chagas’ disease; Trypanosoma cruzi) | Benznidazole, 5-7 mg/kg/day PO in two doses × 60 days | Nifurtimox, 8-10 mg/kg/day PO in three to four doses × 90 days |
| African trypanosomiasis (Trypanosoma brucei gambiense [W. African]; T. b. rhodesiense [E. African] | Pentamidine/suramin/eflornithine/ nifurtimox/melarsoprol | Drug treatment depends on stage of disease (hemolymphatic stage or CNS disease) and on geographic origin (W. or E. African); consult expert guidelines |
* Not specifically approved by the U.S. Food and Drug Administration for use in this infection.
† Relatively ineffective in immunosuppressed patients.
CNS, central nervous system; IM, intramuscularly; IV, intravenously; PO, orally.
Drugs for Leishmaniasis and Trypanosomiasis
For leishmaniasis and trypanosomiasis, selection of drugs is influenced by the geographic location where the infection was acquired and by the clinical features of the infection, particularly its anatomic location. Many of the agents used can be difficult to obtain and for use in the United States, often must be sourced from the Centers for Disease Control and Prevention (CDC). In addition, many of these agents have significant toxicity profiles and complex treatment regimens, so seeking specialist advice is advised.
Amphotericin B
Conventional amphotericin B deoxycholate, typically used as an antifungal agent, has demonstrated efficacy for the treatment of cutaneous leishmaniasis (CL), mucocutaneous leishmaniasis (MCL), and visceral leishmaniasis (VL).1–3 However, toxicity (primarily renal) limits its use, and the better-tolerated liposomal formulation (AmBisome) has emerged as the preferred therapy. Liposomal amphotericin B has been reported to have cure rates greater than or equal to 95% in VL, with Indian VL being the most responsive and Brazilian and Sudanese VL less so.1,4,5 Accumulating case reports and small case series suggest that this agent may also have a role in the treatment of MCL and CL.6–10,11 The U.S. Food and Drug Administration (FDA)-approved regimen for liposomal amphotericin B in VL for immunocompetent patients is 21 mg/kg in seven infusions over a 21-day period (3 mg/kg/day on days 1-5, 14, and 21), although success with a single dose of 10 mg/kg has been reported in India.12,13 The dosing regimens for CL and MCL vary, with some cohorts reporting success with doses of 3 mg/kg/day given for 6 to 10 days.6–8 See Chapter 39 for a full discussion of the uses and toxicities associated with amphotericin.
Antimonials
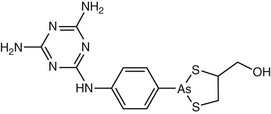
Antimony has been used for medicinal purposes for several centuries, with efficacy for leishmaniasis reported in the early 1900s.14 Pentavalent antimony Sb(V) is used for treatment of leishmaniasis and is available as sodium stibogluconate (sodium antimony gluconate), sold under the brand name Pentostam (GlaxoSmithKline, Brentford, Middlesex, England) (Fig. 41-1), and as meglumine antimoniate, sold under the brand name Glucantime (Sanofi-Aventis, Paris). Generic preparations of both formulations of pentavalent antimony are manufactured in India and China. In the United States, neither meglumine antimoniate nor sodium stibogluconate is commercially available, but sodium stibogluconate can be obtained under an Investigational New Drug protocol through the CDC for civilian physicians and the Walter Reed National Military Medical Center for military physicians.
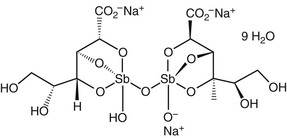
The detailed chemical structure of pentavalent antimony is not fully known, although analysis by electrospray ionization mass spectrometry (ESI-MS) and osmolarity measurements suggest that both meglumine and sodium stibogluconate contain 1 : 1, 1 : 2, 2 : 2, and 2 : 3 Sb(V)-ligand complexes. ESI-MS analysis of meglumine showed negatively-charged 1 : 1 (m/z 364) and 2 : 2 (m/z 765) Sb(V)-meglumine complexes, supporting the predominance of zwitterionic species in solution. ESI-MS measurements of sodium stibogluconate also demonstrate a mixture of oligomeric structures.15
Despite almost a century of use, the mechanism of action of pentavalent antimonials is not completely understood. One model suggests that Sb(V) acts as a prodrug and is reduced to the more active/toxic trivalent form of antimony, Sb(III). Sb(III) affects glucose metabolism, fatty acid beta oxidation, and adenosine triphosphate (ATP) formation.16,17 Other specific targets, such as topoisomerase or trypanothione reductase, have been identified.18,19 Sb(III) also competes with Zn(II) for its binding to the motif CCHC that is a constituent of zinc-finger domains, suggesting that zinc-finger proteins may be targets of antimony.20 A second model holds that Sb(V) has intrinsic antileishmanial activity. Sb(V) has been reported to complex with adenine nucleosides, which might act as an inhibitor of Leishmania purine transporters or interfere with the purine salvage pathway.21 A third model suggests that activation of the host immune system is the major mechanism of action for pentavalent antimonials. Sodium antimony gluconate has been shown to induce nitric oxide synthesis (NO) and reactive oxygen species (ROS), with both ROS and NO involved in parasite killing in the early stage of infection and NO in the late stage.22,23 Sodium antimony gluconate has also been shown to upregulate interferon-γ (IFN-γ) receptors in both Leishmania donovani–infected and –uninfected Th1 cells and in monocytes derived from kala-azar patients treated with sodium antimony gluconate, thus potentially influencing host response by altering IFN-γ responsiveness.24
Although Sb(V) has been successfully used as the first-line drug for leishmaniasis for decades, therapeutic failure to pentavalent antimonials is being increasingly reported. In Bihar State, India, up to 65% of previously untreated patients relapse or fail to respond promptly to therapy because of drug resistance.25 Available evidence indicates that the primary mechanism of resistance is mediated by a reduction in active drug concentration within the parasite. This may occur through decreased uptake, increased efflux, inhibition of drug activation, inactivation of active drug by metabolism or sequestration, or by the development of a bypass pathway. Currently, the exact mechanism of drug resistance is undefined, and no biomarker for antimony susceptibility/resistance has been identified.26,27
Sb(V) can be directly administered into cutaneous lesions for treatment, typically at a dose of 1 to 5 mL sodium stibogluconate injection per session every 3 to 7 days for one to five sessions.28 Systemic therapy is generally administered in a dose of 20 mg/kg/day for 20 to 30 days by intravenous infusion or by intramuscular (IM) injection.29 The maximum licensed dose for sodium stibogluconate is 850 mg per day, but expert opinion and clinical experience support no upper limit to dosing.29 Intravenous administration is preferred over IM injection because of pain associated with injection. Sodium stibogluconate is provided as a 100-mg antimony/mL solution that contains a preservative, m-chlorocresol. The drug is renally excreted, with substantial interindividual variability in clearance. The drug is well absorbed after IM administration, with rapid distribution and a slower elimination half-life of about 10 hours and a first-order absorption rate constant.30,31 Administration to patients with significant renal impairment is not recommended.
Side effects of systemic administration include pancreatitis, hepatitis, arthralgias and myalgias, reactivation of herpes zoster virus, and cardiotoxicity.32–35 To monitor for toxicity, baseline and at least weekly complete blood counts, serum creatinine, serum liver transaminases, serum amylase, and electrocardiograms (ECG) are suggested. Asymptomatic laboratory abnormalities, such as transient elevations of amylase and liver transaminases, are common during therapy but may herald serious toxicity. Marked abnormalities or abnormalities associated with clinical symptoms should prompt a review of the risk/benefit of treatment and a discussion with an expert in the use of antimony. The arthralgias and myalgias associated with antimony use can be severe and persist for weeks after treatment.
Drug interactions with sodium stibogluconate have not been reported, but it is advised that the product be given with caution in patients with cardiovascular disease, a history of ventricular arrhythmias, or other risk factors known to predispose toward QT prolongation, including use of class III antiarrhythmics, such as sotalol and amiodarone. A risk of fatal cardiac arrhythmias has been observed when amphotericin B deoxycholate is administered after sodium stibogluconate during re-treatment of visceral leishmaniasis.36 Sodium stibogluconate should be withheld during pregnancy, and alternative treatments offered.
Triazoles
A variety of triazole agents typically used for treatment of fungal infections have been reported to show activity for treatment of leishmaniasis, most commonly for CL. Fluconazole, itraconazole, and ketoconazole are the drugs with the most data, with varying rates of efficacy.37,38 See Chapter 39 for a full discussion of the uses and toxicities associated with azole drugs.
Miltefosine (Hexadecylphosphocholine, HePC)
Miltefosine (Impavido; Paladin Labs, St. Laurent, Quebec) (Fig. 41-2) is an oral agent originally developed as an antineoplastic agent for treatment of breast cancer and other solid tumors, but its development as an antineoplastic agent was curtailed because of dose-limiting gastrointestinal toxicity. In immunocompetent patients, a 28-day regimen has a reported cure rate of 60% to 80% in MCL and greater than 80% in VL.39–42 Cure rates for CL have ranged widely, perhaps reflecting a species-specific variation in response to therapy.43 The drug is not approved for use in the United States, and must be administered under an Investigational New Drug protocol. Providers seeking to use miltefosine are encouraged to contact the CDC for specific advice. The CDC has also expressed willingness to release miltefosine for treatment of encephalitis caused by free-living ameba (www.cdc.gov/mmwr/preview/mmwrhtml/mm6233a4.htm; posted August 23, 2013). Also see Chapter 275.
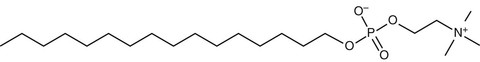
Miltefosine is a member of the alkylphosphocholine class of drugs, which are phosphocholine esters of aliphatic long-chain alcohols. Miltefosine is considered an inhibitor of the AKT protein (also known as protein kinase B [PKB]), which is an important protein within the phosphatidylinositol-3-kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) intracellular signaling pathway, a pathway that is essential for cell survival.44,45
Miltefosine is active in vitro against both promastigotes and amastigotes of various species of Leishmania.46 The mechanism of action is only partly understood and is inferred from data in tumor cell lines where alkyl-lysophospholipids can trigger programmed cell death (apoptosis).47,48 Presumably, miltefosine moves across cell membranes via inward translocation via a transporter. A Leishmania P-type ATPase gene, belonging to the aminophospholipid translocase subfamily, termed LdMT (L. donovani Miltefosine Transporter), has been cloned. LdMT is expressed in the plasma membrane of Leishmania parasites, where it mediates translocation of phospholipids across the plasma membrane.49 The mechanism by which miltefosine induces apoptosis is unclear but may involve the inhibition of the synthesis of phosphatidyl choline, an essential element in the synthesis and integrity of cellular membranes and a source of signaling molecules.44,45
Resistance of Leishmania parasites to miltefosine appears to be related to decreased drug accumulation. Resistant lines achieve low drug levels by two independent mechanisms: either by increasing drug efflux, mediated by the overexpression of the ATP-binding cassette (ABC) transporter P-glycoprotein, or by decreasing drug uptake, which is achieved by the inactivation of any one of the two proteins known to be responsible for the miltefosine uptake, the miltefosine transporter LdMT and its beta subunit LdRos3.50
Miltefosine is administered for 28 days, at a dose of 2.5 mg/kg/day, up to 150 mg/day. Oral bioavailability in humans has not been reported, but the absolute bioavailability in rats and dogs is 82% and 95%, respectively.51 The drug has an extremely long second and terminal elimination half-life of 30.9 days, with drug detected at least 5 months post-treatment.52 Drug interactions are believed to be unlikely. Nausea, vomiting, and diarrhea have been consistently reported, and it is recommended that the drug be taken three times a day with meals to decrease gastrointestinal side effects. Elevated serum creatinine levels are frequently reported, although severe nephrotoxicity is rare. Mild elevations of transaminase levels (both alanine aminotransferase and aspartate aminotransferase) often occur during the first week of treatment.39,53 In a cohort of soldiers treated with miltefosine, 70% were unable to fulfill daily military exercises, and 62% reported temporary diminished ejaculate volume.54 Miltefosine is teratogenic and, thus, administration is contraindicated during pregnancy. Because of the drug’s long half-life, it is recommended that females of childbearing potential use adequate contraception during and for at least 4 months after the standard 28-day treatment regimen.55 Miltefosine is not recommended for use in breast-feeding women.
Paromomycin
Paromomycin (aminosidine) is a legacy off-patent aminoglycoside antibiotic. It is the only aminoglycoside antibiotic with activity against Leishmania species and, in addition, has activity against some protozoa and cestodes.56 Its antileishmanial properties were recognized in 1961.57 Efficacy for treatment of VL has been reported.58,59 Some studies have shown that the combination of paromomycin and sodium stibogluconate is more effective than monotherapy with either drug.60–62 Data supporting the use of injectable paromomycin for the treatment of CL are modest and suggest limited efficacy.63,64 Topical paromomycin, formulated as an ointment with various additional components, has been reported as effective treatment of CL with mixed results.65 A recent randomized study conducted in Tunisia reported that a topical cream containing 15% paromomycin was more effective than placebo in curing patients with CL secondary to Leishmania major.65a Intravenous paromomycin is not available for use in the United States. The drug can be administered orally for clearance of Entamoeba histolytica cysts and for treatment of Dientamoeba fragilis infection.
The leishmanicidal activity of paromomycin is poorly understood, with activity proposed to be mediated through inhibition of parasite metabolism and mitochondrial respiration.66,67 Resistance to paromomycin is readily induced in vitro and appears to result from a decrease in drug uptake. In an in vitro model, the acquisition of resistance by L. donovani did not lead to loss of infectivity and was stable in the absence of continuing drug pressure.68 Paromomycin resistance has been induced in L. major, Leishmania tropica, and in Leishmania aethiopica isolated from patients with diffuse CL after 60 days of treatment with paromomycin; parasites were three to five times less sensitive to the drug than those isolated before treatment.69–71
Paromomycin sulfate is administered at a dose of 11 to 20 mg/kg IM daily for 10 to 21 days. As expected, the important aminoglycoside class adverse drug reactions are renal, cochlear, and vestibular toxicity. The rates of adverse reactions reported in clinical trials have been low, although audiometric studies were not performed in most trials. Insufficient data are available regarding the use of paromomycin in pregnant women, although the drug is thought to be safe during lactation, provided the mother and infant have normal renal function.56 It shows some activity in human cryptosporidiosis,56 with most studies reporting on activity in the setting of human immunodeficiency virus (HIV) infection.72–75 However, available data indicate that the drug alone is not curative. It also has useful activity as an agent to clear cyst carriage of E. histolytica, with a small clinical trial indicting that it is more effective than diloxanide for this indication.76 The dose for this indication and for treatment of D. fragilis infection is 25 to 35 mg/kg/day orally in three divided doses for 7 days.
Pentamidine Isethionate
Pentamidine isethionate (4,4′-[pentane-1,5-diylbis(oxy)]dibenzenecarboximidamide; Nebupent) is an aromatic diamidine that is used to treat the hemolymphatic form (stage 1 disease) of both East and West African trypanosomiasis. It also is a second-line drug for prophylaxis and treatment of Pneumocystis pneumonia77 and is also used for treatment of leishmaniasis.78,79 In addition, pentamidine has activity against Acanthamoeba species and Balamuthia mandrillaris.80,81
Although the precise mechanism of action of pentamidine has not been determined, it is known that pentamidine affects a wide range of microbial processes, including interaction with trypanosomal kinetoplast DNA; interference with polyamine synthesis by decreasing the activity of ornithine decarboxylase; and inhibition of RNA polymerase, ribosomal function, and the synthesis of nucleic acids and proteins.
Intramuscular pentamidine is well absorbed, highly tissue bound, and is excreted slowly over several weeks. Renal secretion is minimal. The elimination half-life is 12 days,82 and it can be detected in plasma up to 8 months after a single dose.83 No steady-state plasma concentration is attained in persons given daily injections; the result is extensive accumulation of pentamidine in tissues, primarily the liver, kidney, adrenal glands, and spleen. Pentamidine does not penetrate the central nervous system (CNS) well, reaching only 0.5% to 0.8% of plasma concentrations after a course of treatment.84 Thus, it should never be used to treat patients with the CNS form of trypanosomiasis (stage 2 disease).
Resistance to pentamidine has been found in human African trypanosomes that are also resistant to melarsoprol. Recent evidence suggests that the unconventional aquaglyceroporin AQP2 renders cells sensitive to both melarsoprol and pentamidine and that loss of AQP2 function could explain cases of innate and acquired pentamidine-melarsoprol cross-resistance.85
Dosing of pentamidine is not generally modified in patients with renal or liver failure. There are no data regarding the pharmacokinetics of pentamidine in obese patients. In animal models, pentamidine is embryocidal but neither teratogenic nor mutagenic.86 Treatment with pentamidine should be delayed until after the first trimester of pregnancy. The relapse rate in children with trypanosomiasis treated with pentamidine is higher than in adults,87 and a delayed response in children with leishmaniasis treated with pentamidine has been noted.88 These observations suggest that the pharmacokinetics of pentamidine in children differ substantially from those in adults, but there are no experimental data to support this concept.
Sterile abscesses can result from IM injections of pentamidine. Hypotension is seen in roughly 15% of patients given the drug, especially if given intravenously (IV) over less than 1 hour. Pentamidine causes some degree of renal insufficiency, usually mild to moderate and reversible, in up to one third of patients. Electrolyte abnormalities, including hyponatremia, hyperkalemia, hypomagnesemia, and hypocalcemia, are common. Hypoglycemia, resulting from a cytotoxic effect on beta-islet cells and insulin release, is an unpredictable and occasionally lethal complication seen in 15% to 25% of patients, particularly with prolonged therapy, azotemia, or high pentamidine levels.89,90 Pentamidine can cause severe pancreatitis, resulting in diabetes over the long term. Neutropenia has been noted; anemia and thrombocytopenia are less frequent. Nausea or vomiting is seen in up to one half of patients, and abnormalities of liver function tests are also common. A variety of electrocardiographic abnormalities, including torsades de pointes, have been reported. Data regarding interactions between pentamidine and other drugs are lacking.
Pentamidine is a second-line therapy for leishmaniasis, predominantly used for CL. Efficacy varies widely, with cure rates ranging from 35% with L. (Viannia) braziliensis in Peru, to 90% with L. (Viannia) guyanensis in Suriname. Use as secondary prophylaxis for VL in immunocompromised patients has been reported in a small number of patients.91 When used for treatment of CL, the dose is usually 3 mg/kg every other day for 4 doses or 2 mg/kg every other day for 7 days. The dose recommended for treatment of mucosal leishmaniasis is 2 to 4 mg/kg every other day or three times per week for 15 doses, and for VL it is 4 mg/kg every other day or three times per week for 15 to 30 doses. See Chapter 39 for a further discussion of the use and toxicities of pentamidine.
Benznidazole
Benznidazole (N-benzyl-2-nitroimidazole-1-acetamide; Rochagan, Radanil) (Fig. 41-3) is a nitroimidazole derivative that is the drug of choice for treating infections with Trypanosoma cruzi. The mechanism of action of benznidazole is not known. Benznidazole is readily absorbed, highly lipophilic, and extensively metabolized, with only 5% of the dose excreted unchanged in the urine.92,93
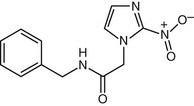
Benznidazole is only available in oral form. The dose and duration of treatment are the same regardless of the stage of the infection. No assays are available commercially to determine blood levels, and no data are available to guide dose adjustments in patients with renal or hepatic insufficiency, pregnancy, or in lactating women. It is recommended that benznidazole not be given to such patients. Resistance to benznidazole has been reported94; efforts to understand the molecular mechanisms underlying resistance are underway.95,95a The clinical significance of resistance is unknown, in large measure because compliance is often an issue, and also because assessment of parasitologic cure after treatment is difficult. No assays for testing for resistance to benznidazole are available, and no data regarding drug-drug interactions have been published.
Adverse effects occur in a substantial proportion of patients treated with benznidazole, but predisposing factors that increase the risk for side effects have not been established.96 Limited data suggest that there is no relationship between benznidazole levels and risk for adverse drug reactions.97 Peripheral neuropathy and rash are the most common. Granulocytopenia can also occur. On occasion, the latter can be severe, and because of this, blood counts should be monitored weekly during the first few weeks of treatment. Adverse effects usually disappear with dose reduction or stopping the drug, although granulocytopenia may take several weeks to resolve. It is noteworthy that patients with Chagas’ disease who undergo cardiac transplantation and who are given benznidazole for management of reactivation of T. cruzi have an increased incidence of malignant tumors.98 However, the incidence of tumors in the general population of patients treated with benznidazole has not been studied.
In the United States benznidazole is available from the CDC Drug Service.
Eflornithine
Eflornithine (difluoromethylornithine, DFMO; Ornidyl) (Fig. 41-4) is a fluorinated analogue of ornithine used for treatment of human African trypanosomiasis (HAT), otherwise known as sleeping sickness. The drug is effective against all stages of West African HAT, caused by Trypanosoma brucei gambiense. However, it lacks activity against East African HAT caused by Trypanosoma brucei rhodesiense.
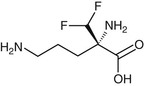
Eflornithine acts as an irreversible suicide inhibitor of ornithine decarboxylase, the first enzyme in the biosynthesis of the polyamines putrescine and spermidine. Interference with polyamine synthesis impairs the ability of the parasite to maintain its redox state and to block reactive oxygen intermediates.95,99 Polyamines also are essential for parasite cell division. Although the drug has a similar effect on humans, there is a selective effect on trypanosomes because they have a relatively low turnover of ornithine decarboxylase and, as a consequence, a more rapid decrease of polyamines with eflornithine treatment.
Eflornithine can be given intravenously or orally, but its bioavailability after oral administration is only 54%. Eflornithine readily crosses the blood-brain barrier, and cerebrospinal fluid (CSF) levels are highest in persons with the most severe CNS involvement of the infection.100 The elimination half-life of eflornithine is 3.3 hours, with greater than 80% excreted in the urine unchanged.101 Given the predominant renal excretion of the drug, dose reduction should occur in patients with renal sufficiency. However, specific data to guide dose adjustments are not available. Dosing need not be adjusted in patients with hepatic dysfunction.
Treatment failures with eflornithine have been reported, with the loss of the gene that encodes the amino acid transporter TbAAT6 being proposed as the molecular basis of this resistance, at least in T. b. gambiense.102,103 The relative lack of activity of eflornithine against T. b. rhodesiense may be based on additional mechanisms.100,104 At the end of a 14-day course of intravenous eflornithine, patients who failed therapy had eflornithine CSF trough concentrations of more than 50 nmol/mL, suggesting parasite resistance to the drug rather than inadequate CSF drug levels as the cause of the failures. The pharmacokinetics of eflornithine in children differ substantially from adults, with mean serum and CSF levels about 60% of levels found in adults; this may explain the higher failure rate in children.105
The most common toxicity of eflornithine is hematologic, with anemia (40%), leukopenia (20% to 30%), and thrombocytopenia (50%) being common. However, these effects are usually mild and without clinical significance. Seizures, associated with higher CSF concentrations, occur more commonly in cases of relapse (12%) than in new cases (4%). An osmotic diarrhea is seen more frequently when eflornithine is given orally. Hearing loss and alopecia have been reported in a few patients. Some patients die during treatment, but this appears to be related to the underlying disease rather than as a result of drug toxicity. In animal models, eflornithine is embryotoxic and induces abortions but is not teratogenic106; anecdotal data suggest that treatment of pregnant women with eflornithine is associated with abortion.107
No data are available concerning interactions between eflornithine and other drugs. However, animal models suggest that eflornithine may be synergistic with other trypanocidal drugs, particularly melarsoprol, because eflornithine reduces the production of trypanothione, a spermidine-glutathione conjugate that is one of the targets of melarsoprol.108,109
Melarsoprol
Melarsoprol (melarsen oxide coupled to 2,3-dimercaptopropinol, mel B, melarsen-BAL; Arsobal) (Fig. 41-5) is an aromatic arsenical that has been used for more than 60 years for treatment of HAT with CNS involvement (stage 2 disease) and for the treatment of hemolymphatic (stage 1 disease) HAT that cannot be effectively treated with suramin or pentamidine. The mechanisms of action of melarsoprol and of drug resistance are complex and have been the subjects of considerable study.94,95,104 The metabolism of trypanothione appears to be a central effect of the drug on the parasite. Melarsoprol interacts with thiol groups of several key proteins, depriving the parasite of its main sulfhydryl antioxidant and inhibiting trypanothione reductase, also depriving the parasite of the essential enzyme system that is responsible for keeping trypanothione reduced.99 Melarsoprol enters the parasite via an adenosine transporter, and resistant strains lack this transport system.110