DNA topoisomerase targeting drugs
Anish Thomas, MBBS, MD  Susan Bates, MD
Susan Bates, MD  William D. Figg Sr, Pharm D
William D. Figg Sr, Pharm D  Yves Pommier, MD, PhD
Yves Pommier, MD, PhD
Overview
DNA topoisomerase targeting drugs are widely used in the treatment of multiple cancers. These drugs act by generating topoisomerase-linked DNA breaks and by blocking the religation of the cleavage complexes. Although topoisomerase targeting drugs have a wide spectrum of activity, structural, pharmacokinetic, and pharmacodynamic characteristics somewhat limit their efficacy and safety. To circumvent these limitations and to further exploit its activity, newer inhibitors- including novel formulations and those that achieve targeted delivery- are being developed. Recently described mechanisms that underlie cellular sensitivity to these agents will enable us to predict the activity of topoisomerase targeting drugs and to precisely select patients who are most likely to benefit. Understanding the genetic variants in drug metabolizing enzymes, transporters, and other proteins that influence the pharmacology of these drugs will allow for further individualizing the use of these drugs.
Introduction
The human genome consists of long double-stranded and helical DNA polymers (46 chromosomes) densely packaged in the cell nucleus (∼1.8 m of DNA squeezed in a nucleus almost 1 million times smaller in diameter). Relaxing DNA supercoiling by topoisomerases is obligatory when the two genomic strands separate for transcription and replication, because the nucleosomal structure of chromatin constrains and generates DNA supercoiling. Moreover, Type II topoisomerases are required at mitosis for the even distribution of the genome between daughter cells following replication. Thus, topoisomerases are ubiquitous and essential for all organisms as they prevent and resolve DNA and RNA entanglements and resolve DNA supercoiling during transcription and replication.
Topoisomerases were named historically with the first topoisomerase being Topo I in Escherichia coli1 and Top1 in mouse.2 Notably, most of the anticancer and antibacterial agents, which are highly specific for their topoisomerase targets, are integral to the anticancer chemotherapeutic armamentarium and were discovered independently and before the term topoisomerase was even coined.
Topoisomerase biology
Human cells contain six topoisomerase genes, with three of the encoded enzymes targeted by anticancer drugs: Top1, Top2α, and Top2β (Table 1).3–5
Table 1 Characteristics of topoisomerases
| Genes | Chromosome | Proteins | Drugs | Mechanism | Polaritya | Main functions |
| TOP1 | 20q12-q13.1 | Top1 100 kDa monomer | Camptothecins Indenos | Swivelling Rotation | 3′-Y | Nuclear supercoiling relaxation |
| TOP1MT | 8q24.3 | Top1mt 70 kDa monomer | None | Mitochondrial supercoiling relaxation | ||
| TOP2A | 17q21-q22 | Top2α 170 kDa dimer | Anthracyclines, Anthracediones Epipodophyllotoxins | Strand passage ATPase | 5′-PY | Decatenation/replication |
| TOP2B | 3p24 | Top2β 180 kDa dimer | Transcription | |||
| TOP3A | 17p12-p11.2 | Top3α 100 kDa monomer | None | Strand passage | 5′-PY | DNA replication with BLMb |
| TOP3B | 22q11.22 | Top3β 100 kDa monomer | RNA topoisomerase |
a Covalent linkage between the catalytic tyrosine and the end of the broken DNA.
b Bloom syndrome, RecQ helicase.
The type of topoisomerase-mediated DNA break is specific of each topoisomerase (Figure 1; Table 1). These catalytic intermediates are referred to as cleavage complexes (Figure 1b, e). The reverse religation reaction is carried out by the attack of the ribose hydroxyl ends toward the tyrosyl-DNA bond.
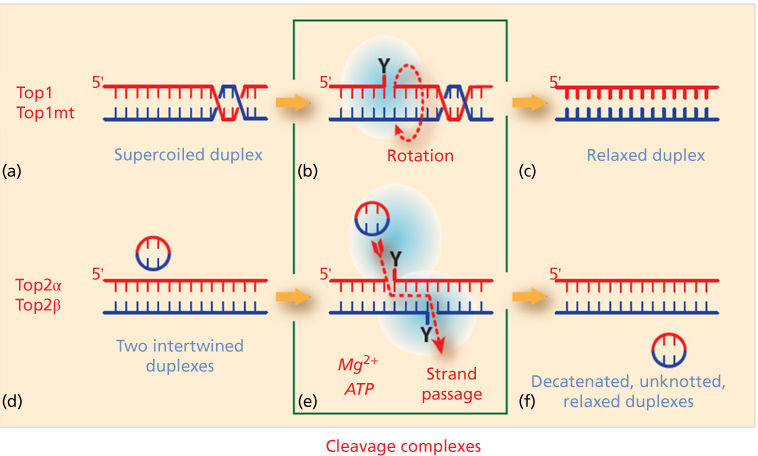
Figure 1 Topoisomerase molecular mechanisms. (a–c) Topoisomerases I (Top1 for nuclear DNA and Top1mt for mitochondrial DNA) relax supercoiled DNA (a) by reversibly cleaving one DNA strand, forming a covalent bond between the enzyme catalytic tyrosine and the 3′-end of the nicked DNA (the Top1 cleavage complex: Top1cc; b). This reaction allows the swiveling of the broken strand around the intact strand. Rapid religation allows the dissociation of Top1 (c). (d–f) Topoisomerases II act on two DNA duplexes. They act as homodimers, cleaving both strands by forming a covalent bond between their catalytic tyrosine and the 5′-end of the DNA break (Top2cc; e). This reaction allows the passage of the intact duplex through the Top2 homodimer (red dotted arrow; e). Top2 inhibitors trap the Top2cc and prevent the normal religation (f). Topoisomerase cleavage complexes (b and d) are the targets of topoisomerase inhibitors.
Top1 (and Top1mt) covalently attaches to the 3′-end of the break, whereas the other topoisomerases (Top2α and β and Top3α and β) attach to the 5′-end of the break Topoisomerases act by cutting and religating the DNA backbone without assistance of nucleases and ligases. Top1 and Top1mt cleave and religate one strand of DNA duplex, whereas Top2α and β cleave and religate both strands with a four base-pair staggered cut (Figure 1). The DNA cutting-religation mechanism is common to all topoisomerases and utilizes an enzyme catalytic tyrosine residue acting as nucleophile and becoming covalently attached to the end of the broken DNA. The polarity of the attachment to the 3′- versus the 5′-ends of the DNA Top3α and β has the opposite polarity, with covalent attachment to the 5′-end of the breaks (Table 1; Figure 1b, e). Topoisomerases are biochemically distinct. Top1 and Top1mt act as monomers in the absence of nucleotide or metal cofactors, whereas Top2α and β act as dimers, requiring ATP and Mg2+ for catalysis. Top3α and β also require Mg2+ but function as monomers without ATP. Notably, the DNA substrates differ for Top3 enzymes. Both Top1 and Top2 process double-stranded DNA, whereas the Top3 substrates need to be single-stranded nucleic acids (DNA for Top3α and RNA for Top3β).
Mechanisms of action
Common mechanism of action and molecular pharmacology of topoisomerase inhibitors: trapping the topoisomerase cleavage complexes by interfacial inhibition
Topoisomerase cleavage complexes are normally highly reversible and therefore transient and hardly detectable in the absence of the topoisomerase inhibitor. This is because the religation of the cleavage complexes is driven by the realignment of the broken ends, which itself is determined by two basic structural features of double-stranded DNA: (1) the stacking of adjacent bases by π–π interactions within each strand, and (2) the pairing of bases across the opposite strands by hydrogen bond interactions (duplex DNA can be viewed as a “powerful molecular zipper”).
The clinical topoisomerase inhibitors act by generating topoisomerase-linked DNA breaks as they block the religation of the cleavage complexes when a single drug molecule tightly binds at the interface of the topoisomerase-DNA cleavage complex. The selectivity and strength of the drug binding is established by (1) the stacking of the drug with the bases flanking the cleavage site and (2) a network of hydrogen bonds with the topoisomerase (Figure 2). As the drug is bound within the cleavage site, it prevents DNA religation by misaligning the DNA end, which is normally required to attack the phosphotyrosyl bond. This mode of binding led to the concept of “interfacial inhibition,” which applies not only to protein–DNA interfaces but also to protein–protein interfaces, as in the case of tubulin and mTOR inhibitors.6 Crystal structures of drug-bound cleavage complexes have firmly established this mechanism for both Top1- and Top2-targeted drugs.6 The structural characteristics of each drug (differences in chemical scaffold and arrangement of hydrogen-bond donors and acceptors) (Figure 3) accounts for the selectivity of each drug for Top1 versus Top2 cleavage complexes, and for the differential DNA sequence selectivity and genomic targeting for different topoisomerase inhibitors within each class.5, 7
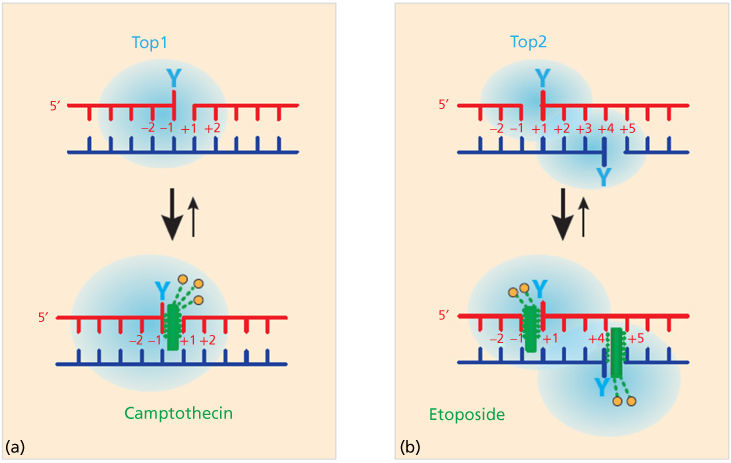
Figure 2 Trapping of topoisomerase cleavage complexes by interfacial inhibition. (a) Binding of camptothecin and non-camptothecin Top1 inhibitors (green rectangle) at the cleavage site generated by the Top1. A single-drug molecule binds reversibly to the Top1cc by stacking against the bases flanking the cleavage site (intercalation) and by a network of hydrogen bonds with Top1 (orange circles and dashed green lines). (b) Same for etoposide with a single-drug molecule (green) intercalated in the cleavage site formed by each Top2 monomer. Hydrogen bonds with Top2 are shown as orange circles and dashed green lines.
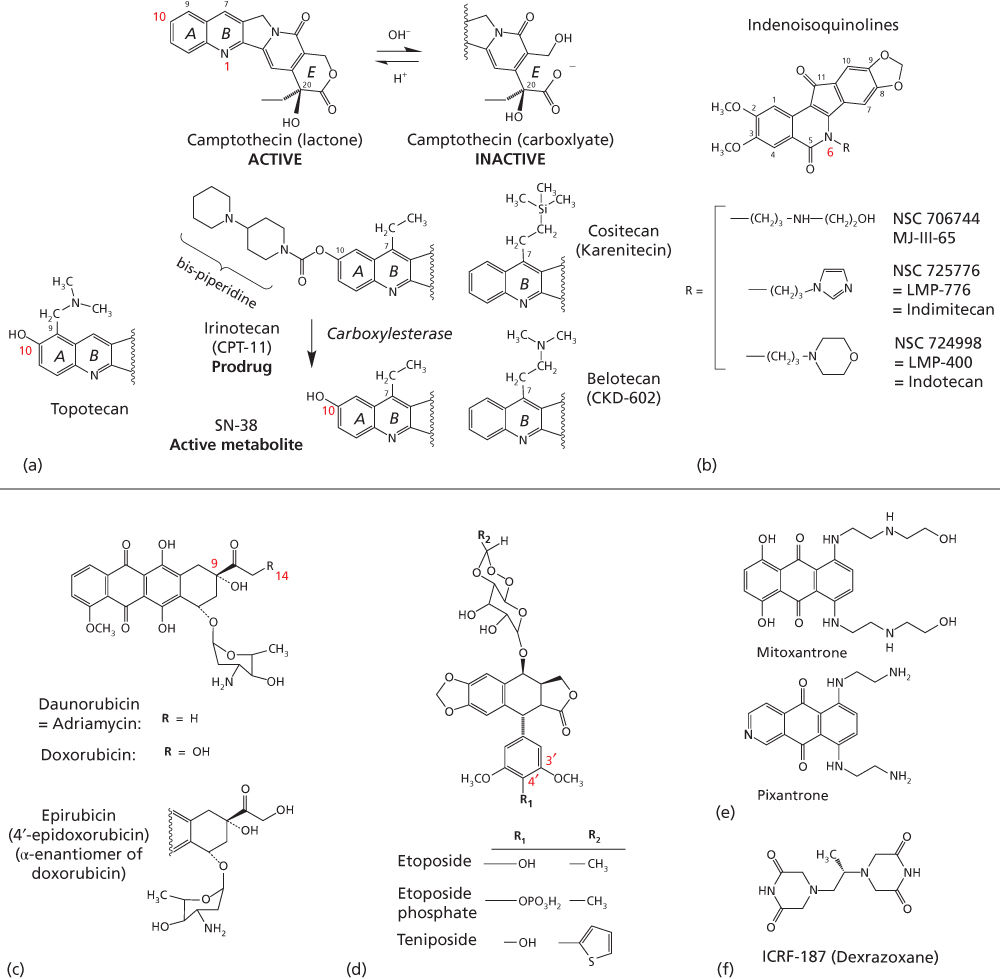
Figure 3 Clinical topoisomerase inhibitors. (a) Camptothecin derivatives are instable at physiological pH, with formation of a carboxylate derivative within minutes. Irinotecan is a prodrug, which needs to be converted to SN-38 to trap Top1cc. (b) Non-camptothecin indenoisoquinoline derivatives in clinical trials. (c) Anthracycline derivatives. (d) Demethylepipodophyllotoxin derivatives. (e) Mitoxantrone. (f) Dexrazoxane, which acts as a catalytic inhibitor of Top2.
It is key to understand that the cytotoxicity of topoisomerase inhibitors is due to the trapping of topoisomerase cleavage complexes as separate from the associated topoisomerase catalytic inhibition. Except in molecularly defined settings, it is the topoisomerase cleavage complexes (the DNA breaks and associated topoisomerase-DNA covalent complexes) that kill cancer cells. This sets apart topoisomerase inhibitors from classical enzyme inhibitors such as antifolates. Indeed, knocking out Top1 renders yeast cells totally immune to camptothecin8 and reduction of enzyme levels renders cancer cells drug resistant.3, 5 Also, mutations of TOP1 and TOP2 that render cells insensitive to the trapping of topoisomerase cleavage complexes confer high resistance to Top1 or Top2 inhibitors. Conversely, amplification of TOP2A, which is on the same locus as HER2, contributes to the selectivity and activity of doxorubicin in breast cancers with amplification at this locus.
The outcome of topoisomerase trapping is determined first by the ability of the cell to repair the cleavage complexes, and then by two common pathways: the apoptotic and the cell cycle checkpoint pathways. Topoisomerase repair pathways are specific to Top1 or Top2 and are discussed in the paragraphs that follow. Defects in pro-apoptotic molecules and excess of anti-apoptotic molecules, which are commonly associated with cancers, confer global resistance to topoisomerase inhibitors, as well as to other anticancer agents. Systematic analyses of cell lines with specific DNA repair alterations revealed that cells deficient in DNA double-strand break repair may be selectively sensitive to topoisomerase inhibitors.9 Genomic analyses recently uncovered a previously unsuspected common pathway that determines cellular sensitivity to both Top1 and Top2 inhibitors (and also other DNA damaging agents, including cisplatin and carboplatin). This novel pathway is centered on the gene SLFN11 (Schlafen 11).10, 11 This finding could be viewed as a paradigm for discovering genomic determinants and signatures to predict response to topoisomerase inhibitors and staging of patients who are most likely to respond to topoisomerase inhibitors.
Molecular pathways specific for cancer cell killing and DNA repair by Top1 inhibitors
Top1cc-targeted drugs (topotecan, irinotecan, and indenoisoquinoline derivatives) kill cancer cells primarily by replication collisions. Indeed, Top1cc’s by themselves are not cytotoxic as they remain reversible even in the presence of drugs that increase their persistence.12 Top1cc’s damage DNA by replication and transcription fork collisions. This explains why cytotoxicity is directly related to drug exposure and why arresting DNA replication protects cells from camptothecin.4 Collisions arise from the fact that the drugs, by slowing down the nicking-closing activity of Top1, uncouple the Top1 from the polymerases and helicases, leading polymerases to collide into Top1cc. Such collisions have two consequences. They generate double-strand breaks (replication and transcription run-off) and irreversible Top1-DNA adducts, known as cleavage complexes. The replication double-strand breaks are repaired by homologous recombination, which explains the hypersensitivity of BRCA-deficient cancer cells to Top1cc-targeted drugs.9 The Top1-covalent complexes can be removed by two pathways, the excision pathway involving tyrosyl-DNA-phosphodiesterase 1 (TDP1) and the endonuclease pathway involving 3′-flap endonucleases such as XPF-ERCC1.13 It is also possible that drug-trapped Top1cc directly generate DNA double-strand breaks when they are within 10 base pairs on opposite strands of the DNA duplex or when they occur next to a preexisting single-strand break on the opposite strand.14 Finally, it is not excluded that topological defects contribute to the cytotoxicity of Top1cc-targeted drugs (accumulation of supercoils15) and formation of alternative structures such as R-loops16 and reversed replication forks.17
Molecular pathways specific for cancer cell killing and DNA repair by Top2 inhibitors
Contrary to Top1 inhibitors, Top2 inhibitors can kill cancer cells independently of DNA replication fork collisions. The collision mechanism in the case of Top2cc-targeted drugs involves transcription and proteolysis of both Top2 and RNA polymerase II, leading to DNA double-strand breaks by disruption of the Top2 dimer. Alternatively, the Top2 homodimer interface could be disjoined by mechanical tension. It is important to note that 90% of Top2cc trapped by etoposide are not concerted and therefore consist in single-strand breaks, which is different from doxorubicin, which produces a majority of Top2-mediated DNA double-strand breaks.4, 5 Finally, it is not excluded that topological defects resulting from Top2 sequestration by the drug-induced cleavage complexes could contribute to the cytotoxicity of Top2cc-targeted drugs. Such topological defects would include persistent DNA knots and catenanes, potentially leading to chromosome breaks during mitosis. Top2 covalent complexes are removed by TDP213 in conjunction with the end-joining pathway [Ku, DNA-dependent protein kinase (DNA-PK), ligase IV, and XRCC4].9, 18
The anticancer activity of intercalating Top2 inhibitors (anthracyclines and anthracenediones) extends beyond the trapping of Top2 cleavage complexes
Some Top2 inhibitors are also potent DNA intercalating agents; this is the case of the anthracyclines (Figure 3c) and mitoxantrone (Figure 3e). Consequently, anthracyclines and mitoxantrone affect Top2cc in two ways: at low drug concentrations, they trap Top2cc, whereas at higher concentrations (>5 μM), high levels of intercalation outside of the Top2cc suppress the binding of Top2 to DNA and thereby inhibit the formation of Top2cc. As a result, the concentration-response curve of doxorubicin is “bell-shaped.”4, 19 Intercalation of anthracylines also destabilizes chromatin structure with nucleosome eviction.20 Another property of the anthracyclines is the generation of oxygen radicals due to their quinone structure.21
Topoisomerase I inhibitors
Topotecan
Camptothecin and its derivatives were found to have a high degree of in vitro antitumor activity in the 1960s, which was then confirmed in early clinical trials.22–24 Clinical development was limited by unpredictable toxicities, including severe myelosuppression and diarrhea, and an incomplete understanding of the mechanism of antitumor activity. Topotecan has since been shown to be a specific inhibitor of the nuclear enzyme Top125 Following this, topotecan has been developed for a number of clinical indications (Table 2).
Table 2 DNA topoisomerase targeting drugs
| Drug class | Drugs | Year of FDA approval | Approved indication |
| Camptothecin | Irinotecan | 1996 | Colorectal cancer |
| Topotecan | 1996 | Cervical cancer, ovarian cancer, small cell lung cancer | |
| Anthracycline | Doxorubicin | 1974 | Breast cancer, acute lymphoblastic leukemia, acute myeloid leukemia, Wilms tumor, neuroblastoma, soft tissue and bone sarcomas, ovarian cancer, transitional cell bladder carcinoma, thyroid carcinoma, gastric carcinoma, Hodgkin lymphoma, non-Hodgkin lymphoma, bronchogenic carcinoma |
| Pegylated liposomal doxorubicin | 1999 | AIDS-related Kaposi sarcoma, multiple myeloma, ovarian cancer | |
| Daunorubicin | 1979 | Acute lymphocytic leukemia, acute myeloid leukemia | |
| ldarubicin | 1990 | Acute myeloid leukemia | |
| Anthraquinone | Mitoxantrone | 1987 | Acute nonlymphocytic leukemias, advanced hormone-refractory prostate cancer |
| Podophyllotoxin | Etoposide | 1984 | Small cell lung cancer, testicular cancer |
| Teniposide | 1992 | Acute lymphoblastic leukemia |
Clinical pharmacology
Topotecan (9-dimethylaminomethyl-10-hydroxycamptothecin) (Figure 3a) is a semisynthetic analog of camptothecin, an alkaloid derived from the oriental tree Camptotheca acuminata. The side chain at the 9-position of the A-ring (Figure 3a), provides water solubility. Lactone topotecan, the active form of the drug, is unstable as it is rapidly and reversibly converted to the open-ring carboxylate in a pH-dependent reaction12 (Figure 3a). At neutral pH, the open-ring form predominates.26
Topotecan pharmacokinetics fit a two-compartment model with a terminal half-life of 2–3 h. The plasma concentrations increase linearly with increasing doses, but do not show evidence of accumulation on the 30-min infusion X 5-day schedule.27 Binding of topotecan to plasma proteins is about 35%.27
Topotecan lactone is widely distributed, with a steady-state mean volume of distribution of 75 L/m2. Topotecan is predominantly eliminated in the urine after its conversion to the carboxylate species. Although large interindividual variability exists, about 30% of the administered dose is excreted unchanged in urine.27
Renal dysfunction decreases topotecan plasma clearance, such that mild dysfunction (creatinine clearance 40–60 mL/min) and moderate dysfunction (creatinine clearance 20–39 mL/min), lead to reductions to 67% and 34% of normal values, respectively.28 Dose adjustments are therefore recommended in case of renal impairment. Liver impairment does not influence topotecan clearance and doses do not need to be adjusted for patients with hepatic dysfunction.29
Although the typical route of administration is IV, an oral formulation is available. The oral formulation of topotecan was aimed at maintaining sufficiently prolonged drug exposures, which are known to produce highest antitumor efficacy in vitro and in vivo,12 without the need for infusion pumps.30 The approved oral dose is 2.3 mg/m2 once daily for 5 consecutive days repeated every 21 days. The oral bioavailability of topotecan is approximately 35%.27 The low bioavailability may be caused by hydrolysis of topotecan lactone in the gut, yielding the poorly absorbed open-ring form. Oral topotecan has shown clinical activity comparable to the IV dose in small cell lung cancer (SCLC).31
Clinical use
Topotecan monotherapy is approved in the United States and Europe for patients with metastatic ovarian cancer patients after failure of initial or subsequent chemotherapy, and in patients with relapsed SCLC. It is also approved in combination with cisplatin for recurrent or resistant (stage IVB) cervical cancer.32
Topotecan has shown clinical activity in other tumor types, including pediatric medulloblastoma,33 non-SCLC,34 myelodysplastic syndrome, chronic myelomonocytic leukemia,35 Ewing’s sarcoma,36 rhabdomyosarcoma,37 and multiple myeloma.38 In addition to the combination with cisplatin approved in cervical cancer, combinations of Top1 inhibitors with other antitumor agents have been pursued. Synergistic and additive combinations have been observed in preclinical models, including with histone deacetylase inhibitors and PARP inhibitors, and a number of drug combination strategies have been investigated in patients.39, 40 Topotecan diffuses to the spinal fluid and can be considered for treating brain metastases.41
Schedule of administration
Various schedules of topotecan have been evaluated in phase I studies. The FDA (Food and Drug Administration)-recommended dose of topotecan in recurrent ovarian cancer and SCLC is 1.5 mg/m2 by IV infusion over 30 min on days 1 to 5 of a 21-day cycle.
Despite the high incidence of grade 3 or 4 myelosuppression associated with this dose and schedule of topotecan, it remains the standard of care. To mitigate this toxicity, multiple dose and schedule variations have been evaluated in phase I and II trials.42, 43 In ovarian cancer, weekly topotecan (4 mg/m2/week administered on days 1, 8, and 15) has been reported to have efficacy comparable to the standard schedule, but with less toxicity.43 The weekly schedule has not been directly compared with the 5-day schedule in SCLC.31 In cervical cancer, topotecan is approved in combination with cisplatin. In this setting, the recommended dose of topotecan is 0.75 mg/m2 IV over 30 min on days 1, 2, and 3 with cisplatin 50 mg/m2 IV on day 1 repeated every 21 days.44
Toxicity
The dose-limiting toxicity of topotecan is bone marrow suppression, primarily neutropenia, but thrombocytopenia and anemia may also occur. At a dose of 1.5 mg/m2/day for 5 days, topotecan produces an 80% to 90% decrease in white blood cell count at nadir after the first cycle of therapy. The degree of neutropenia has been correlated with the area under the curve (AUC) of the intact lactone or total drug concentrations.45 Topotecan should not be administered to patients with baseline neutrophils <1500/mm3 or platelets <100,000/mm3.
As discussed above, renal dysfunction decreases topotecan clearance and increases toxicity. Additional factors associated with greater hematologic toxicity include advance age and prior therapy, including platinum administration (especially carboplatin), radiation therapy.46 In high-risk patients receiving topotecan for 5 consecutive days, the incidence of severe myelosuppression may be mitigated by reduction of the topotecan dose to 1.0 or 1.25 mg/m2/day. Hematopoietic growth factors, transfusions, and schedule adjustments may also help manage myelosuppression. Alternative schedules of 3-day, weekly dosing or oral administration are associated with less myelosuppression.47
Other toxicities of topotecan less common than bone marrow suppression include rash, fever, fatigue, nausea and vomiting, mucositis, and elevated serum transaminases.48 Most nonhematological side effects are generally manageable. Diarrhea is uncommon with IV administration, but has been reported with oral topotecan. Rare, but life- threatening nonhematologic toxicities includes interstitial lung disease and neutropenic colitis.
Irinotecan
Irinotecan is a potent water-soluble camptothecin derivative.
Clinical pharmacology
Irinotecan (CPT-11) is the prodrug of the potent 7-ethyl-10-hydroxy analog of camptothecin (SN-38) (Figure 3a).45 It contains a bispiperidine substituent at C-10, to confer water solubility for parenteral administration. Irinotecan undergoes extensive metabolic conversion by esterases to its active metabolite SN-38 (Figure 3a). Irinotecan prodrug is activated by carboxylesterases to SN-38, the biologically active compound. This explains why irinotecan is orders of magnitude less active than SN-38 in in vitro cytotoxicity assays.49, 50
Irinotecan peak plasma concentration and AUC increase proportionally with the administered dose, suggestive of linear pharmacokinetics. The plasma AUC of SN-38 is only 2% to 8% of irinotecan, indicating that only a small fraction of irinotecan is converted to SN-38, its active form. Both irinotecan and SN-38 exist in the active lactone form and the inactive carboxylate form. Similar to topotecan, both forms are in a pH-dependent equilibrium.12
Irinotecan plasma concentrations decline in a multiexponential manner after IV infusion. The mean terminal elimination half-life of irinotecan is 6 to 12 h and that of SN-38 10 to 20 h, both much longer than that of topotecan. The relatively large percentage of the lactone form of both irinotecan and SN-38, which persists in plasma after drug administration, is attributable to the preferential binding of the lactone forms to albumin.26 SN-38 is 95% bound to plasma proteins compared to approximately 50% for irinotecan.45
Unlike topotecan, irinotecan elimination occurs predominantly by biliary excretion.45 Renal excretion of SN-38 and irinotecan represents only a fraction of the administered dose. The 1A1 isoform of uridine 5′-diphospho-glucuronosyl transferase (UGT1A1; encoded by the UGT1A1 gene) mediates glucuronidation of SN-38 to the inactive metabolite SN-38G (see section titled “Pharmacogenomics”). There is wide interpatient variability in UGT1A1 enzyme activity related to UGT1A1 gene polymorphisms.51, 52 In addition, polymorphisms of this enzyme are associated with conditions causing familial hyperbilirubinemia such as Crigler–Najjar syndromes and Gilbert’s disease.53 UGT1A1 polymorphisms can significantly alter the metabolism of irinotecan and thereby impact its toxicity in individual patients, and dose reduction is recommended for patients bearing particular variants of UGT1A1. Irinotecan is also inactivated by CYP3A4-mediated oxidative metabolism.54 The impact of polymorphic variants is discussed in more detail in the subsequent sections.
Clinical use
Irinotecan is approved in the United States and Europe for first-line therapy in combination with 5-fluorouracil and leucovorin for metastatic colorectal cancers. It is also approved for metastatic colorectal cancer in patients whose disease has recurred or progressed following fluorouracil-based therapy.49, 55
Irinotecan monotherapy or combinations with other agents have shown clinical activity in diverse tumor types, including extensive-stage SCLC,56 squamous cell carcinoma of the cervix,57 recurrent glioblastoma,58 gastric cancer, esophageal cancer,59 non-SCLC,60 pancreatic cancer,61 rhabdomyosarcoma,62 and ovarian cancer.63
Schedule of administration
Monotherapy with irinotecan is usually administered at 125 mg/m2 IV infusion over 90 min on days 1, 8, 15, and 22 followed by 2-week rest (6-week treatment cycles). An alternative dosing regimen is 350 mg/m2 given as a 60-min IV infusion once every 3 weeks. In patients with 5-fluorouracil-refractory, metastatic colorectal cancer, the weekly and once every 3-week schedule demonstrated similar efficacy and quality of life. The regimen of once every 3 weeks was associated with a significantly lower incidence of severe diarrhea.64
In combination with 5-fluorouracil and leucovorin, irinotecan is used at 125 mg/m2 IV infusion over 90 min on days 1, 8, 15, and 22 (6-week treatment cycles). An alternative dosing regimen is 180 mg/m2 over 90 min on days 1, 15, and 29 (6-week treatment cycles).
Toxicity
Diarrhea and myelosuppression are the most concerning toxicities of irinotecan. Late-onset diarrhea occurs more than 24 h after irinotecan. When prolonged, it can lead to life-threatening dehydration and electrolyte imbalance.65 Grade 3 or 4 late-onset diarrhea occurs in about a third of patients receiving weekly dosing. The median time to the onset of late diarrhea was 5 days with 3-week dosing and 11 days with weekly dosing.49, 55 Free intestinal luminal SN-38, either from bile or SN-38G deconjugation is responsible for late-onset diarrhea. SN-38 induces direct mucosal damage with resultant water and electrolyte malabsorption and mucous hypersecretion.66 Late diarrhea should be treated without delay with loperamide, and fluid and electrolytes as needed. Antibiotic therapy is warranted if the patient develops ileus, fever, or severe neutropenia. Predisposing factors for late-onset diarrhea include increasing age, poor performance status, and prior abdominopelvic radiotherapy.67 A reduced starting dose should be considered in patients with risk factors. Treatment-related diarrhea should have fully resolved before initiating the next course of treatment. Dose reductions are recommended for grades 3 and 4 diarrhea. Various measures to reduce its severity have been studied, but none have an established role in practice.68 In 2005, the FDA approved a diagnostic test for the UGT1A1 variant allele *28, which has been associated with reduced expression leading to impaired clearance and increased toxicity.69 However, other variant alleles that impair expression of the enzyme are also known and impact clearance of drugs requiring glucuronidation (discussed in greater detail below).
Less commonly, irinotecan can also cause early-onset diarrhea during or within 24 h of the infusion. It is usually transient and only infrequently severe. It is attributed to a cholinergic syndrome mediated by anticholinesterase activity of irinotecan.70 This cholinergic syndrome tends to occur at higher irinotecan dose levels at peak irinotecan plasma levels. Other cholinergic symptoms including abdominal cramping, rhinitis, tearing, and increased salivation may occur. The mean duration of symptoms is 30 min and usually responds rapidly to atropine.
Table 3 Investigational Top1 inhibitors in clinical development
| Drugs/drug class | Comments | |
| Camptothecin analogs | Belotecan | Water-soluble camptothecin analog |
| Cositecan | Lipophilic, semisynthetic camptothecin | |
| Lipotecan | Lactone ring modified to increase antitumor potency | |
| Simmitecan | Ester prodrug of the Top1 inhibitor chimmitecan | |
| Targeted camptothecin delivery | Liposomal/nanoparticle/PEGylated formulations | |
| MM-398 Onivyde | Nanoliposomal formulation of irinotecan | |
| CPX-1 | Liposome-encapsulated formulation of Irinotecan and floxuridine | |
| Firtecan pegol | Pegylated formulation of SN-38 | |
| CRLX101 | Cyclodextrin-based polymer conjugate of camptothecin | |
| Antibody-drug conjugates | ||
| Sacituzumab govitecan | Antibody drug conjugate [antibody that binds to trophoblast cell-surface antigen (TROP-2) and SN-38] | |
| Camptothecin conjugates | ||
| HA-irinotecan | Irinotecan complexed with hyaluronic acid | |
| Etirinotecan pegol (NKTR-102) | Irinotecan bound to polyethylene glycol core by a biodegradable linker | |
| Non-camptothecin Top1 inhibitors | Indenoisoquinoline derivatives | Chemical stability, stable DNA-TOP1 trapping, no affected by drug efflux pumps |
Other common toxicities observed with irinotecan are nausea, vomiting, abdominal pain, constipation, anorexia, asthenia, fever, decrease in body weight, and alopecia. Rare, but life-threatening nonhematological toxicities include interstitial lung disease and hypersensitivity reactions. Risk factors for pulmonary toxicity include preexisting lung disease, use of pulmonary toxic medications, radiation, and use of colony-stimulating factors. Patients with risk factors should be monitored for respiratory symptoms while on irinotecan and for several weeks after treatment.71
Camptothecin analogs and non-camptothecin Top1 inhibitors
As discussed above, camptothecin derivatives have several well-established limitations. They are inactivated within minutes at physiological pH by lactone E ring opening (Figure 3a). This results in a loss of antitumor activity since the lactone form is essential for antitumor activity. Other limitations include fast reversal of the trapped DNA-Top1 cleavage complex following drug removal and drug resistance mediated by ATP-binding cassette (ABC) transporters.12
Several newer derivatives of camptothecin currently in clinical development aim to mitigate some of the shortcomings of camptothecins and their derivatives (Table 3). Camptothecin analogs, derived from modifications to the parent drug, have been an area of active research.72 Belotecan hydrochloride, a water-soluble camptothecin analog is in clinical development for SCLC and ovarian cancer.73 Cositecan (Karenitecin) (Figure 3a) is an orally active, highly lipophilic, semisynthetic camptothecin, under development for ovarian cancer. Due to its lipophilicity, cositecan exhibits enhanced tissue penetration and bioavailability compared to water-soluble camptothecins and are less sensitive to drug resistance.
Several approaches have been taken to stabilize the lactone ring of camptothecins without interfering with their antitumor activity.19 Indenoisoquinolines are non-camptothecin inhibitors of Top1, but with better chemical stability, producing stable DNA-Top1 cleavage complexes. They also have a preference for unique DNA cleavage sites, compared with their camptothecin counterparts and have demonstrated activity against camptothecin-resistant cell lines and produce DNA protein cross-links, which are resistant to reversal. They also show less or no resistance to cells overexpressing the ABC transporters, ATP-binding cassette subfamily G member 2 (ABCG2), and multidrug resistance (MDR)-1.74 Other approaches to address E-ring instability include the conversion of the E ring to a five-membered ring, which completely stabilizes the drug.75
Targeted delivery of Top1 inhibitors
Several approaches are being explored to improve the targeted delivery, and tumor localization of camptothecins. Broadly they involve (1) liposomal or nanoparticle formulation to increase plasma half-life and tumor localization, (2) antibody conjugation for targeted delivery, and (3) conjugation to agents to improve pharmacokinetic properties and exposure.
Liposomal/nanoparticle formulations
Liposomes are microscopic phospholipid spheres with an aqueous core.76 Due to their biphasic character, they can act as carriers for both lipophilic and hydrophilic drugs; hydrophilic drugs tend to be entrapped in the core, whereas the hydrophobic ones are entrapped within the lipid bilayers. Stable liposomal encapsulation of camptothecin minimizes its conversion to the inactive carboxylate form by reducing the direct exposure of the drug to physiologic pH and prolonging the residence time of the lactone active drug. PEGylation increases the size and molecular weight of conjugated biomolecules.77 PEGylated molecules show increased half-life, decreased plasma clearance, and different biodistribution compared with non-PEGylated counterparts. PEGylation of liposomes further improves the stability and circulation time. Polymeric nanoparticles are drug carriers that are designed to have defined size and surface properties to favor drug deposition and retention in tumors.78 These formulations are thought to improve the “passive” targeting of tumors, through a process known as the enhanced permeation retention effect, wherein macromolecules penetrate and are trapped in tumor tissue due to the abnormally leaky vasculature of tumors.79
MM-398 (nal-IRI) is a nanoliposomal encapsulation of irinotecan sucrosofate. In preclinical studies, MM-398 improved the pharmacokinetics and tumor biodistribution of both irinotecan and SN-38 when compared with the free form of the drug, with less accumulation in organs responsible for dose-limiting toxicities.80 MM-398 has shown clinical activity in combination with 5-fluorouracil and leucovorin in metastatic pancreatic cancer patients who have previously received gemcitabine-based therapy.81
CRLX101 is a nanoparticle therapeutic comprised of camptothecin covalently conjugated to a linear, cyclodextrin-polyethylene glycol copolymer.72 CRLX101 self-assembles in solution into nanoparticles with solubility increases of over 1000-fold compared with the parent drug camptothecin. CRLX101 is currently being evaluated in patients with renal, ovarian, and rectal cancers.
Antibody drug conjugates
Antibody drug conjugates (ADCs) generated by conjugating camptothecins to monoclonal antibodies are being investigated to facilitate targeted drug delivery to tumors. Sacituzumab govitecan is a humanized antibody against trophoblast cell-surface antigen (TROP-2) conjugated by a pH-sensitive linker to SN-38.82 TROP-2 is a type I transmembrane, calcium-transducing, protein expressed by many epithelial cancers, but with limited normal tissue expression. The antibody internalizes selectively into cancer cells following binding to TROP-2 encoded by the TACSTD2 gene. Sacituzumab govitecan is being investigated in triple-negative breast cancer and SCLC.
Camptothecin conjugates
Etirinotecan pegol is a long-acting topoisomerase-I inhibitor that consists of irinotecan conjugated to a polyethylene glycol core by a biodegradable linker. The linker slowly hydrolyses in vivo to form SN38, the active moiety. The drug is designed to provide prolonged continuous exposure to SN38 while reducing the toxicities associated with excessively high irinotecan and SN38 plasma concentrations. Etirinotecan pegol at a dose of 145 mg/m2 administered IV every 21 days is being studied in a phase III trial in women with advanced breast cancer.83 Hyaluronic acid-irinotecan is an IV formulation of irinotecan conjugated with hyaluronic acid.84 Hyaluronic acid selectively binds cancer cells via CD44, thereby enhancing membrane fluidity and potentially reducing side effects.
Top2 inhibitors
Top2 targeting drugs used in clinic can be divided into intercalating and non-intercalating poisons. The intercalators are chemically diverse, and include doxorubicin and other anthracyclines, and anthracenediones (Figure 3c, e). Non-intercalating Top2 poisons include the epipodophyllotoxins, etoposide and teniposide (Figure 3d; Table 2).
Anthracyclines
Anthracycline antibiotics, originally isolated from fermentation products of Streptomyces peucetius, were shown to have antineoplastic activity decades ago, before topoisomerase enzymes were identified. It was not until 1984 that the anthracyclines were found to inhibit Top2.85 Doxorubicin, also known as adriamycin and daunorubicin, represent first-generation anthracyclines; epirubicin and idarubicin are second-generation compounds aimed at reducing cardiotoxicity and drug efflux by ABC transporters. Anthracyclines have wide-ranging activity against human cancers and are used extensively in the curative, adjuvant, and palliative settings, both as single agents and in combination regimens.
Doxorubicin is the most commonly used anthracycline (Figure 3c). The anthracycline ring is lipophilic, but the presence of abundant hydroxyl groups adjacent to the amino sugar produces an amphoteric molecule. Doxorubicin binds to cell membranes, including cardiolipin as well as plasma proteins. Daunomycin differs from doxorubicin by a single hydroxyl group on carbon-14 (Figure 3c), but has a distinct spectrum of antitumor activity. Idarubicin is a semisynthetic derivative of daunomycin (4-demethoxy daunorubicin) without the 4-methoxy group (Figure 3
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


