Disorders of the thyroid in the newborn and infant
EMBRYOLOGY, PHYSIOLOGY, AND PHYSIOPATHOLOGY
Development of the Thyrotropic Axis
Placental Transfer of Iodine, T4, TRH, Antithyroid Drugs, and Immunoglobulins
Maturation of Thyroid Hormone Synthesis and Secretion
Maturation of Thyroid Hormone Metabolism and Transport
Extrauterine Thyroid Adaptation
Introduction
The management of the fetus, newborn, and infant with disorders of thyroid function has benefited greatly from our ability, since the 1970s, to measure hormones in minute amounts of biologic fluids. Specifically, biochemical screening of newborns for congenital hypothyroidism (CH) is now routine throughout the industrialized world and has resulted in the disappearance of the overt intellectual disability that affected 8% to 28% of patients with this condition in the prescreening era.1 More recently, the molecular basis of several monogenic disorders of thyroid development and function has been deciphered. However, most cases of abnormal thyroid gland development in humans, the most common cause of CH, are unexplained and constitute one of the remaining mysteries of thyroid physiopathology.2 On the other hand, progress in ultrasound techniques has led to an increasing number of goiters being identified in the fetus in utero, which raises the possibility of prenatal treatment via the mother’s amniotic fluid in highly selected cases.3 This chapter reviews thyroid disorders in the fetus, newborn, and infant from a developmental, molecular, and clinical perspective.
Embryology, physiology, and physiopathology
Development of the thyrotropic axis
As for all organs, anatomic development of the hypothalamic-pituitary-thyroid system occurs during the first trimester of gestation. The human embryonic forebrain and hypothalamus begin to differentiate by 3 weeks of gestation under the influence of a series of homeodomain proteins or transcription factors. Immunoreactive thyrotropin-releasing hormone (TRH) becomes detectable in human embryonic hypothalami by 8 to 9 weeks of postconceptional age and is also produced by the fetal gut and pancreas.4,5
Anatomically, the pituitary gland develops from two ectodermal anlagen: a neural component from the floor of the primitive forebrain and Rathke’s pouch from the primitive oral cavity. The latter is visible by 5 weeks, evolving to a morphologically mature pituitary gland by 14 to 15 weeks. The pituitary-portal blood vessels are present by this time and mature further through 30 to 35 weeks. A wide spectrum of congenital malformations collectively named “midline defects,” including holoprosencephaly and septo-optic dysplasia, may be associated with central hypothyroidism and other anterior pituitary deficiencies.6 The molecular mechanisms underlying these malformations have been identified in some cases.7 Within the pituitary itself, PROP-1 and PIT-1 are terminal factors in the differentiation cascade of pituitary cells and PIT-1 or PROP-1 deficiency results in profound defects in growth hormone, prolactin, and thyroid-stimulating hormone (TSH) secretion as well as age-dependent pituitary hypoplasia.8–10
The human thyroid gland develops from a median anlage derived from the primitive pharyngeal floor and from paired lateral anlagen from the fourth pharyngeal pouches. The long-held belief that the lateral anlagen were the only source of calcitonin-producing cells has been challenged by the observation that sublingual thyroids that are derived exclusively from the median anlage contain calcitonin mRNA and protein.11 Conversely, thyroid follicular cells can differentiate within the lateral anlagen, as illustrated by histologic observations12 as well as by patients in whom the only thyroid tissue is a lateral ectopy.13 Both the median and lateral structures are visible by day 16 to 17 of gestation; by 50 days they have fused and the thyroid gland has migrated to its definitive location in the anterior neck. The thyroglossal duct, from the foramen cecum to the final location of the thyroid, may persist and is constituted of degenerated thyroid follicular cells. Within the thyroid gland, iodine concentration, TSH receptors, thyroglobulin, and thyroid peroxidase mRNA and protein can be demonstrated by 70 days.14
Thyroid embryogenesis is dependent on the expression of a programmed sequence of homeobox and transcription factors, including thyroid transcription factors-1 and -2 (TTF-1 or TiTF-1, also known as NK2 homeobox 1 -NKX2-1- and TTF-2, also known as Forkhead box E1 -FOXE-1) and paired box gene 8 (PAX8).15 Embryonic stem cells in which NKX2-1 and PAX 8 are overexpressed form fully differentiated thyroid cells and will form the typical follicular architecture under the influence of TSH.16 In keeping with this concept, mice with a naturally occurring mutation inactivating the TSH receptor have a disorganized follicular architecture,17 and this is also probably the case in humans with similar mutations in which a high serum thyroglobulin in spite of profound thyroid hypoplasia suggests leaky follicles.18 In newborn mice, biallelic inactivation of Nkx2.1 results in absence of both pituitary and thyroid glands, with complete absence of both thyroid follicular cells and of calcitonin-producing C cells,19 whereas that of PAX8 results in a small thyroid gland composed almost exclusively of C cells.20 FOXE-1 null mouse embryos have either an absent thyroid or an ectopic sublingual gland, but all newborn pups have athyreosis in addition to cleft palate.21 Mutations in the homologous genes, however, account for at most 2% of cases of thyroid dysgenesis in humans.22
Placental transfer of iodine, T4, TRH, antithyroid drugs, and immunoglobulins
Iodine is an essential component of thyroid hormones. In this chapter, the term iodine is used to designate both iodine itself (I2) and iodide (I–). The human placenta expresses the sodium–iodine symporter throughout gestation,23 which explains why the mother’s iodine status is reflected in the fetus (Figure 7-1) . If the mother’s iodine intake is suboptimal, the fetal thyroid cannot constitute appropriate stores of iodine and fetal hypothyroidism may ensue. Worldwide, inadequate maternal iodine intake leading to irreversible consequences, known as endemic cretinism, remains a major public health problem. Prevention of this condition by supplying the mother with adequate iodine is one of the best and most important examples of considering the fetus as a patient and of treating this patient through its mother.24,25
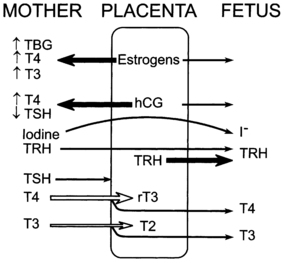
FIGURE 7-1  The placental role in thyroid metabolism during human pregnancy. The placenta produces estrogens and a human chorionic gonadotropin (hCG), which increase maternal TBG levels and stimulate maternal thyroid hormone production, respectively. Both activities tend to increase maternal T4 and T3 concentrations and inhibit maternal TSH secretion. Iodide and TRH readily cross the placenta. In addition, the placenta synthesizes TRH. The placenta is impermeable to TSH and only partially permeable to T4 and T3. Placental type 3 iodothyronine monodeiodinase enzymes degrade T4 to reverse T3 and T3 to 3,3’ T2. The placenta is also permeable to the thiourea drugs used to treat maternal Graves disease.
The placental role in thyroid metabolism during human pregnancy. The placenta produces estrogens and a human chorionic gonadotropin (hCG), which increase maternal TBG levels and stimulate maternal thyroid hormone production, respectively. Both activities tend to increase maternal T4 and T3 concentrations and inhibit maternal TSH secretion. Iodide and TRH readily cross the placenta. In addition, the placenta synthesizes TRH. The placenta is impermeable to TSH and only partially permeable to T4 and T3. Placental type 3 iodothyronine monodeiodinase enzymes degrade T4 to reverse T3 and T3 to 3,3’ T2. The placenta is also permeable to the thiourea drugs used to treat maternal Graves disease.
In contrast to iodine, thyroxine (T4) was for a long time thought not to cross the placenta in substantial amounts.26 However, T4 is detectable in human embryonic tissues before the onset of fetal thyroid function and must, therefore, be of maternal origin.27 Later in gestation, the transfer of T4 from mother to fetus must continue, because the concentration in cord blood from neonates with complete absence of thyroid function is 30% to 50% of that of normal neonates.28 More recently, it was shown that normal neonates born to a mother who has chronically higher T4 concentrations due to a mutation inactivating the thyroid hormone receptor have lower birth weight and TSH concentrations at newborn screening than those born to normal mothers.29 Taken together, these data indicate that maternal T4 must cross the placenta in physiologically relevant amounts throughout gestation.
That this transplacental transfer of T4 from mother to fetus might be of clinical relevance was suggested by the severe developmental delay observed in an infant with central hypothyroidism caused by a maternally inherited heterozygous mutation inactivating PIT1, and whose equally hypothyroid mother had stopped thyroxine treatment at midpregnancy.30 At the population level, children born to mothers who have low T4 concentrations during pregnancy have been reported to have lower IQ than children born to mothers with normal circulating concentrations of thyroid hormones.31–33 However, in a randomized trial, developmental outcome was similar in the offspring of T4- and placebo-treated women who had either elevated TSH concentrations (> 97.5 percentile) or lower circulating T4 concentrations (< 2.5 percentile) when treatment was begun toward the end of the first trimester.34 Moreover, in two recent case series of women with severe hypothyroidism diagnosed during pregnancy but corrected by the third trimester, the intellectual outcome of the offspring was normal.35,36 Thus, universal screening for thyroid dysfunction during pregnancy remains questionable at this time. On the other hand, 85% of women who are already receiving T4 therapy require an increase in dose of about 30% to 50% during pregnancy because of the estradiol-induced increase in serum thyroxine-binding globulin (TBG).37
The transplacental transfer of T4 is not always sufficient to prevent the development of fetal goiter if the fetus has severe thyroid dyshormonogenesis. Fetal goiters may be big enough to interfere with the flow of amniotic fluid into the airways, causing progressive hydramnios and eventual lung hypoplasia. In such occurrences, levothyroxine can be injected into the amniotic fluid, which the fetus then swallows, leading to a decrease in the size of the fetal thyroid and in the degree of hydramnios.38 The injection of thyroxine into the umbilical vein, which carries an even higher risk of triggering premature labor or fetal loss than amniocentesis, should be restricted to fetuses with a goiter that continues to increase in spite of repeated intra-amniotic injections. Invasive and potentially risky procedures should not be undertaken to protect the brain of affected fetuses: indeed, the fetal brain is, to a large extent, protected from the deleterious effect of hypothyroidism through up-regulation of brain type 2 deiodinase, which converts the prohormone T4 into its biologically active derivative, T3.39 This likely accounts for the observation that even in CH with delayed bone maturation at diagnosis (indicating a prenatal onset), the intellectual outcome is within normal limits if continuous and adequate treatment is instituted shortly after birth.40 Thus, the in utero treatment of fetal hypothyroidism should only be considered in exceptional circumstances, such as for goiter causing progressive hydramnios. Although the identification of a goiter by prenatal ultrasound may be increasing,41 it remains rare and even direct examination at birth often fails to detect goiters that are obvious on scintigraphy. Goiters can also be observed in fetuses borne by women with Graves disease if they are overtreated with antithyroid drugs, which readily cross the placenta. Reducing the dose of the antithyroid medication given to the mother should decrease the size of the fetal thyroid in such circumstances42 (Figure 7-2).
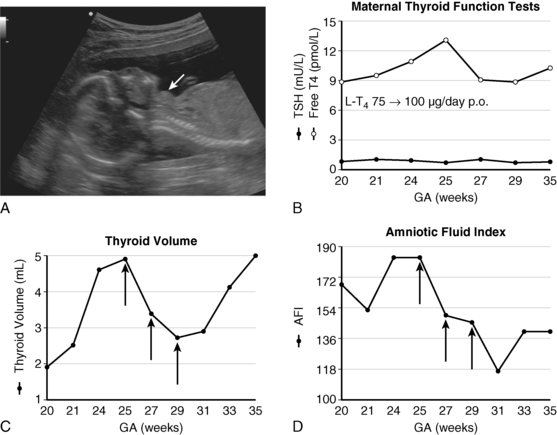
FIGURE 7-2  A, Fetal goiter discovered at a routine ultrasound at 19 weeks (white arrow). The mother was euthyroid and had no autoantibodies. Cordocentesis showed a serum TSH of 90 mU/L. B, initial management was through oral treatment of the mother with increasing doses of levothyroxine; this resulted in increased maternal free T4 concentrations, but presumably not enough to cross the placenta in sufficient amounts to prevent the progression of the fetal goiter (panel C) and the development of hydramnios (panel D). C, Effect of 3 intra-amniotic injections of 200 μg of levothyroxine (black arrows) on fetal thyroid size. D, Effect of three intra-amniotic injections of 200 μg of levothyroxine (black arrows) on amniotic fluid index. At birth, cord serum TSH was 224 mU/L and thyroglobulin was low at 3.55 μg/L; thyroglobulin deficiency was confirmed by molecular genetic analyses. Treatment with 50 μg of levothyroxine orally from day 1 has allowed this child, now age 4 years, to have normal intellectual development. (Adapted and updated from Stoppa-Vaucher S, Francoeur D, Grignon A, et al. [2010]. Non-immune goiter and hypothyroidism in a 19-week fetus: a plea for conservative treatment. J Pediatr 156:1026-1029.)
A, Fetal goiter discovered at a routine ultrasound at 19 weeks (white arrow). The mother was euthyroid and had no autoantibodies. Cordocentesis showed a serum TSH of 90 mU/L. B, initial management was through oral treatment of the mother with increasing doses of levothyroxine; this resulted in increased maternal free T4 concentrations, but presumably not enough to cross the placenta in sufficient amounts to prevent the progression of the fetal goiter (panel C) and the development of hydramnios (panel D). C, Effect of 3 intra-amniotic injections of 200 μg of levothyroxine (black arrows) on fetal thyroid size. D, Effect of three intra-amniotic injections of 200 μg of levothyroxine (black arrows) on amniotic fluid index. At birth, cord serum TSH was 224 mU/L and thyroglobulin was low at 3.55 μg/L; thyroglobulin deficiency was confirmed by molecular genetic analyses. Treatment with 50 μg of levothyroxine orally from day 1 has allowed this child, now age 4 years, to have normal intellectual development. (Adapted and updated from Stoppa-Vaucher S, Francoeur D, Grignon A, et al. [2010]. Non-immune goiter and hypothyroidism in a 19-week fetus: a plea for conservative treatment. J Pediatr 156:1026-1029.)
Although the placenta produces a pro-TRH molecule, TRH concentrations in the maternal circulation are very low and thus have little effect on fetal thyroid function. However, TRH, with its tripeptide structure (the smallest of the hypothalamic hypophysiotropic peptides), crosses the placenta readily and, when injected to the mother, increases thyroid hormone concentrations in the fetus. Because thyroid hormones stimulate fetal lung maturation, maternal TRH treatment to decrease neonatal respiratory distress syndrome has been attempted, but without successful outcome.43
Because immunoglobulins of the IgG type cross the placenta, transient fetal/neonatal hyperthyroidism from transplacental transfer of TSH-receptor activating antibodies can occur in women with past or present Graves disease. On the other hand, when pregnant women are overtreated with antithyroid drugs, which also cross the placenta readily, their fetus may develop goitrous hypothyroidism, as noted previously. However, in the Québec database for neonatal thyroid screening, only one case out of 30,000 births is attributable to this mechanism (unpublished observations). Lastly, transient neonatal hypothyroidism from materno-fetal transfer of TSH-receptor–blocking antibodies can also occur, but only accounts for 2% of cases of neonatal hypothyroidism identified by screening44; hence, for babies born to women with Hashimoto thyroiditis, a specific screening strategy is not required. However, thyroid function tests are often ordered clinically in newborns whose mother has a history of thyroid disease and, if abnormal, require special consideration of optimal approach to therapy. For example, hypothyroidism at birth caused by treatment of maternal Graves disease with antithyroid medication may only require observation, in the expectation that the effects of the drugs will dissipate over a few days; hyperthyroidism may follow, albeit exceptionally. For hypothyroidism or hyperthyroidism resulting respectively from maternal TSH receptor blocking or stimulating antibodies, treatment will be required as these effects may last several months.45
Maturation of thyroid hormone synthesis and secretion
Maturation of thyroid function in the fetus reflects changes at the level of the hypothalamus, pituitary, and thyroid. Serum TRH concentrations are relatively high in the human fetus, because it is produced at both hypothalamic and extrahypothalamic sites and because of the TRH-degrading activity of fetal blood is low.46 Fetal serum TSH increases from a low concentration at 18 to 20 weeks to a peak value of approximately 7 to 10 mU/L at term. After delivery, in response to exposure to the cold extrauterine environment, there is an acute release of TSH with mean serum concentrations peaking at 30 minutes at approximately 70 mU/L. The subsequent increase in serum T4 concentration immediately after birth is TSH dependent.
Only free thyroid hormones enter cells, and hormones bound to serum TBG and other transport proteins are not available to tissues. In addition, T4 is a prohormone and it is T3 that is biologically active to exert intracellular effects, so that deiodination of T4 is essential for tissue euthyroidism. Both serum transport proteins and intracellular deiodination change during development. As previously mentioned, the fetal thyroid gland is capable of iodine concentration and iodothyronine synthesis as early as 70 days of gestation, a reflection of a sharp increase in the expression of the sodium-iodine symporter and of the appearance of a follicular architecture.47 Starting at 18 to 20 weeks, total T4 and TBG concentrations in fetal serum increase steadily until the final weeks of pregnancy.
The study of free T4 concentrations in fetal/neonatal blood has been hampered by the relative inadequacy of the commercially available immunoassay systems for measurements in these samples.48 The fetal serum T3 concentration remains low until 30 weeks due to two factors: first, the low activities of type 1 iodothyronine monodeiodinase result in relatively low rates of T4 to T3 conversion in fetal tissues; second, active type 3 monodeiodinase in placenta and selected fetal tissues degrades T3 to T2. After 30 weeks, serum T3 increases slowly until birth. This prenatal increase in serum T3 is due to progressive maturation of liver type 1 deiodinase activity increasing hepatic conversion of T4 to T3, and to decreased placental T3 degradation. Postnatally, T3 and T4 serum concentrations increase two- to sixfold within the first few hours, peaking on the second day of life. These levels then gradually decline to levels characteristic of infancy over the first 4 to 5 weeks of life (Figures 7-3 and 7-4).
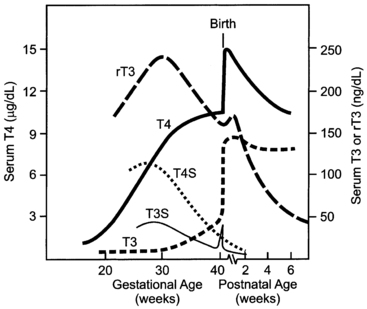
FIGURE 7-3  The pattern of change in fetal and neonatal thyroid function parameters during pregnancy and extrauterine adaptation. Fetal serum thyroxine (T4) concentrations begin to increase at midgestation and increase progressively thereafter to term. This increase is due largely to the increase in thyroxine-binding globulin concentration, but free T4 concentrations (not shown) also increase progressively between 20 and 40 weeks. T4 in the fetus is metabolized predominantly to inactive reverse triiodothyronine (rT3) and sulfated analogs (T4S, T3S). Monodeiodination of T4 to active triiodothyronine (T3) increases at about 30 weeks to levels approximating 50 ng/dL at term. The TSH surge (not shown), which peaks at 25 to 30 minutes after extrauterine exposure, stimulates thyroidal T4 and T3 secretion. Neonatal T4 and T3 peak at 2 to 3 days. Serum T3 concentrations remain at higher postnatal levels because of the increased T4-to-T3 conversion mediated by increased type 1 iodothyronine deiodinase activity in newborn tissues.
The pattern of change in fetal and neonatal thyroid function parameters during pregnancy and extrauterine adaptation. Fetal serum thyroxine (T4) concentrations begin to increase at midgestation and increase progressively thereafter to term. This increase is due largely to the increase in thyroxine-binding globulin concentration, but free T4 concentrations (not shown) also increase progressively between 20 and 40 weeks. T4 in the fetus is metabolized predominantly to inactive reverse triiodothyronine (rT3) and sulfated analogs (T4S, T3S). Monodeiodination of T4 to active triiodothyronine (T3) increases at about 30 weeks to levels approximating 50 ng/dL at term. The TSH surge (not shown), which peaks at 25 to 30 minutes after extrauterine exposure, stimulates thyroidal T4 and T3 secretion. Neonatal T4 and T3 peak at 2 to 3 days. Serum T3 concentrations remain at higher postnatal levels because of the increased T4-to-T3 conversion mediated by increased type 1 iodothyronine deiodinase activity in newborn tissues.
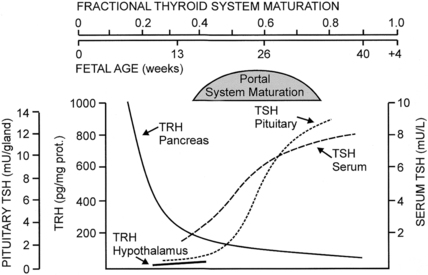
FIGURE 7-4  Changes in fetal TRH and TSH levels in pancreas, hypothalamus, serum, and pituitary during human gestation. Hypothalamic TRH concentrations increase progressively after midgestation, but the pattern of change has not been documented in the human fetus. (Reproduced with permission from Fisher DA, Polk DH [1994]. Development of the fetal thyroid system. In Thorburn GD, Harding R [eds.], Textbook of fetal physiology. Oxford, UK: Oxford University Press, 359-368.)
Changes in fetal TRH and TSH levels in pancreas, hypothalamus, serum, and pituitary during human gestation. Hypothalamic TRH concentrations increase progressively after midgestation, but the pattern of change has not been documented in the human fetus. (Reproduced with permission from Fisher DA, Polk DH [1994]. Development of the fetal thyroid system. In Thorburn GD, Harding R [eds.], Textbook of fetal physiology. Oxford, UK: Oxford University Press, 359-368.)
In the human, the fetal thyroid gland grows and its production increases under the influence of the increasing serum TSH level during the second half of gestation, as illustrated by the severely atrophic and hypofunctional glands observed in newborns with mutations that inactivate either the β-subunit of TSH49 or the TSH receptor.50 On the other hand, the maturation of the negative feedback control system appears to occur earlier than previously thought, as an elevated TSH in serum obtained at cordocentesis can be seen in fetuses with primary hypothyroidism as early as 18 weeks.51
Thyroid function in the premature infant (before 30 to 32 weeks) is characterized by low circulating concentrations of T4 and free T4, a normal or low concentration of TSH, and a normal or prolonged TSH response to TRH, suggesting a degree of relative hypothalamic (tertiary) hypothyroidism. In summary, fetal thyroid hormone secretion results from increasing hypothalamic TRH secretion that stimulates the secretion of TSH from the pituitary and increased thyroid follicular cell sensitivity to TSH. In turn, this process is regulated by increasing pituitary sensitivity to inhibition by thyroid hormone of TSH release. The marked cold-stimulated TRH-TSH surge at birth is associated with a marked increase in T4 secretion and free T4 concentration with a new equilibrium reached by 1 to 2 months. During infancy and childhood, there is a progressive decrease in T4 secretion rate (based on a μg/kg/day) correlating with a decreasing metabolic rate.52
Maturation of thyroid hormone metabolism and transport
Two types of outer ring iodothyronine monodeiodinases have been described.53 Type 1 deiodinase (predominantly expressed in liver, kidney, and thyroid) is a high-Km enzyme inhibited by propylthiouracil and stimulated by thyroid hormone. Type 2 deiodinase (predominantly located in brain, pituitary, placenta, skeletal muscle, heart, thyroid, and brown adipose tissue) is a low-Km enzyme insensitive to propylthiouracil and inhibited by thyroid hormone. Types 1 and 2 deiodinases contribute to circulating T3 production, whereas type 2 acts to increase local tissue levels of T3 as well. An inner ring deiodinase (type 3 deiodinase) has been characterized in most fetal tissues, including placenta. This enzyme system catalyzes the conversion of T4 to rT3 and T3 to diiodothyronine. All three deiodinase enzymes are selenoproteins (Figure 7-5).
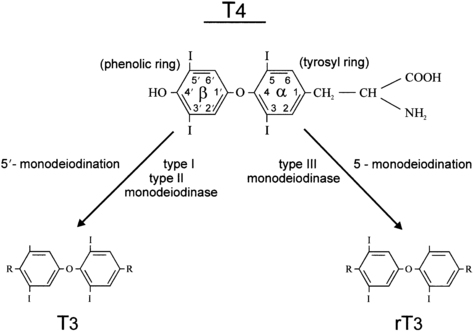
FIGURE 7-5  The deiodination of thyroxine by types 1, 2, and 3 iodothyronine monodeiodinase enzymes. The type 1 enzyme is also capable of inner ring monodeiodination, particularly of the sulfated conjugates (not shown).
The deiodination of thyroxine by types 1, 2, and 3 iodothyronine monodeiodinase enzymes. The type 1 enzyme is also capable of inner ring monodeiodination, particularly of the sulfated conjugates (not shown).
Sulfated iodothyronines are the major thyroid hormone metabolites circulating in the fetus.54 Sulfokinase enzymes are present early in fetal life, and sulfation of the phenolic hydroxyl group of the iodothyronine molecule may be a normal prerequisite step for monodeiodination. The sulfated iodothyronines are preferred substrates for the type 1 deiodinase, and concentrations are high in fetal serum in part because of low type 1 deiodinase activity. However, increased production of sulfated metabolites is also involved. There is evidence that T3S has biologic activity (i.e., suppresses TSH in vivo), suggesting that it can be de-sulfated by one or more tissue sulfatase enzymes. The low production rates and low levels of T3 metabolites and the high ratio of inactive to active metabolites suggest that fetal thyroid hormone metabolism is largely oriented to inactivating T4, presumably to avoid tissue thermogenesis and to potentiate the anabolic state of the rapidly growing fetus. This is mediated by early activation of type 3 monodeiodinase, inactivation of type 1 monodeiodinase, and augmented iodothyronine sulfation.
The developmental expression of type 2 deiodinase in brain and other tissues provides for local T3 supply to specific tissues (particularly in the event of T4 deficiency) and helps guarantee provision of T3 during gestation, when brain development is thyroid hormone dependent.39
Extrauterine thyroid adaptation
At the time of parturition, the neonate must rapidly convert from the fetal state of predominant thyroid hormone inactivation to a state of relative thyroidal hyperactivity. During the first hours after birth, there is an acute increase in circulating T4 and T3 levels. This is due to the abrupt increase in hypothalamic TRH stimulating increased pituitary TSH secretion and, in turn, thyroid hormone secretion. The cold-stimulated TRH-TSH surge is short lived and mean TSH concentrations decrease progressively to normal infant levels by 3 to 5 days, but the serum-free T4 level remains elevated for several weeks.55
Serum T3 levels increase in response to the TSH surge, because of stimulation of thyroidal T3 secretion and of increased hepatic type 1 deiodinase activity. Placental separation decreases T3 deiodination (to inactive T2), contributing to the early postnatal increase in serum T3 concentration. The type 2 deiodinase activity in brown adipose tissue increases during the last weeks of gestation to potentiate catecholamine-stimulated brown adipose tissue thermogenesis, thereby contributing to the maintenance of the body temperature of the neonate.53
Thyroid hormone actions
Evidence suggests that all thyroid-sensitive cell populations express iodothyronine membrane transporters. These belong to several families of integrin, organic anion, amino acid, and monocarboxylate solute carriers. The importance of these transporters is highlighted by the role of mutations inactivating human monocarboxylate transporter 8 (MCT8) in an X-linked syndrome of severe psychomotor retardation (previously called the Allan-Herndon-Dudley syndrome) combined with mild abnormalities of thyroid function, characterized by high T3, low T4, and normal or high TSH.56,57 MCT8 is thought to play a role in the entry of T3 into neurons, after deiodination of T4 to T3 in neighboring astrocytes. In addition, MCT8 is involved in the transfer of T3 across the blood-brain barrier. Lastly, the abnormalities in thyroid hormone levels and TSH are also due to the effect of MCT8 on deiodination.58
In humans, the specific roles of TRα and TRβ are illustrated by the phenotypes observed in patients with inactivating mutations in the corresponding genes. The syndrome of thyroid hormone resistance initially described in 1967 was later found to be generally due to mutations inactivating TRβ that occur either de novo or are inherited in an autosomal dominant fashion; however, in some patients with thyroid hormone resistance, TRβ is normal and the molecular defect remains elusive.59 Mutations in TRα have been described, occurring de novo in one patient60 and transmitted from father to daughter in one pedigree.61
Thyroid hormone-programmed development of fetal tissues requires the interaction of local tissue monodeiodinase 1 and 2, TRs, thyroid receptor coactivators, and thyroid-responsive genes. In most responsive tissues, the timing of maturation events is controlled by the TRs acting as a molecular switch. In the absence of T3, the unliganded receptor recruits corepressors, thereby repressing gene transcription. Local tissue maturation events are stimulated by the coincident availability of T3, liganded T3 receptor, T3-mediated receptor exchange of corepressors with coactivators, and activation of responsive gene transcription.62
In the human fetus, low levels of TR binding have been detected in brain tissue at 10 weeks gestational age—and liver, heart, and lung TR binding is observed at 16 to 18 weeks. TR levels in human fetal cerebral cortex and cerebellum increase markedly during the second and third trimesters.63 Information is limited regarding the timing of the appearance of thyroid hormone tissue effects in the human fetus.
The birth length of the athyroid human neonate is within normal limits: the linear growth of the human fetus is programmed independently of thyroid hormones by a complex interplay of genetic, nutritional, and hormonal factors, as well by the mechanical uterine constraint.64 However, 50% to 60% of athyroid newborns manifest delayed epiphyseal maturation and have large fontanelles.65 At birth, severe CH may be suspected in the presence of a large anterior fontanelle, a persistent posterior fontanelle at term with diastasis of the parietal bones, macroglossia and umbilical hernia; delayed meconium emission, feeding difficulties, and prolonged jaundice may be noted soon thereafter.66 However, the classic clinical manifestations of CH appear progressively during the early months of life. These include soft-tissue myxedema, a slow linear growth, and retarded central nervous system (CNS) development.64 The normal IQ in most athyroid infants treated early thanks to newborn screening1 seems attributable to the low but significant levels of maternal thyroxine derived from placental transfer, shown in humans,28 and the up-regulation of type 2 monodeiodinase in fetal brain tissue in the face of low fetal serum thyroxine concentrations, shown in rats.39
Postnatal thermogenesis is mediated via the brown adipose tissue prominent in subscapular and perirenal areas in the mammalian fetus and neonate. Heat production in brown adipose tissue is stimulated by catecholamines via β-adrenergic receptors and is thyroid hormone dependent. The uncoupling protein thermogenin unique to brown adipose tissue is located on the inner mitochondrial membrane and uncouples phosphorylation by dissipating the proton gradient created by the mitochondrial respiratory chain. The type 2 monodeiodinase in brown adipose tissue mediates local T4 to T3 conversion. Full thermogenin expression in brown adipose tissue requires both catecholamine and T3 stimulation.67 Brown adipose tissue matures progressively in the fetus but remains thermoneutral until stimulated by catecholamines in the perinatal period. Brown adipose tissue thermogenesis is immature in small premature infants, and brown adipose tissue mass decreases in the neonatal period in full-term infants as the capacity for nonshivering thermogenesis develops in other tissues.68 Uncoupling protein-2 is found in many tissues, but it does not appear to be regulated by β-adrenergic agonists or thyroid hormone. Uncoupling protein-3 is expressed in muscle and white adipose tissue as well as brown adipose tissue. Muscle uncoupling protein-3 is regulated by β3-adrenergic stimulation and thyroid hormone and presumably contributes to nonshivering thermogenesis in humans. mRNA levels for uncoupling protein-3 are also regulated by dexamethasone, leptin, and starvation, but the regulation differs in brown adipose tissue and muscle. Starvation increases muscle and decreases brown adipose tissue uncoupling protein-3, suggesting that muscle serves a larger role in thermoregulation during starvation.69
The critical role of thyroid hormones in CNS maturation has long been recognized. Nervous system development involves neurogenesis, gliogenesis, neural cell migration, neuronal differentiation, dendritic and axonal growth, synaptogenesis, myelination, and neurotransmitter synthesis. Thyroid hormones have been shown to stimulate a number of developmentally regulated nervous tissue genes, but the role of these factors in the CNS developmental program remains undefined. Available evidence suggests that deficiency or excess of thyroid hormones alters the timing or synchronization of the CNS developmental program, presumably by initiating critical gene actions or other genetic CNS maturation events.70
There is increasing evidence for a critical period for thyroid-dependent brain maturation in utero. Early maternal hypothyroxinemia in the rat alters histogenesis and cerebral cortical architecture of the progeny.71 As noted earlier, maternal hypothyroxinemia in humans has been reported to be associated with IQ reduction in offspring,31–33 but association does not mean causation as shown by the negative results of the randomized controlled trial of T4 treatment.34 In contrast, iodine supplementation in pregnant women in geographic areas of severe iodine deficiency improves developmental outcome of the offspring if given before the third trimester of gestation.24 The reasons for the discrepancy between the effect of iodine in iodine-deficient women and the lack of effect of T4 in hypothyroid women are unknown. Studies of the dose and timing of thyroid hormone therapy in infants with CH suggest a second critical period of thyroid hormone action during the very early neonatal period,72 but the period of CNS thyroid hormone dependency extends further, to at least 2 years of age.73
Congenital hypothyroidism
Newborn screening
CH screening tests are usually carried out in dried blood spot samples collected via skin puncture, although a few programs use cord serum.74–76 The historical controversy between the proponents of using total T477 or TSH78 as the primary test was related in part to the greater precision of T4 measurements around the cutoff value, but this is no longer an issue. Because primary hypothyroidism is at least 10-fold more common than central hypothyroidism and because only 19% of cases of central hypothyroidism have T4 below the cutoff,6 primary TSH screening makes more sense. About half of the states in the United States and the Netherlands still use a primary T4 strategy, but, in effect, all are screening for elevated serum TSH concentrations as the secondary screening test. In the Dutch program, for instance, TSH is measured in almost half of the population: all those with a T4 below 0.8 SD and all infants born with a weight ≤ 2.5 kg or a gestational age of ≤ 36 weeks.79 Conversely, the Québec program uses T4 to sort cases with borderline TSH elevation (15 to 30 mU/L) as shown on the algorithm in Figure 7-6.
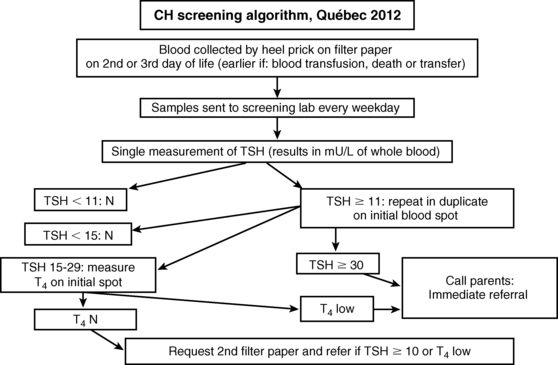

With the trend toward earlier discharge of newborns in the 1990s, the minimal age at which TSH screening should be performed was assessed: in the first 24 hours, 9% of the population had a TSH above 15 mU/L (a reflection of the neonatal surge), but this percentage dropped precipitously to only 0.2% to 0.3% from days 2 to 5. Thus, any sample collected after 24 hours is acceptable.80
A more recent controversy relates to the threshold value for a significant TSH elevation, with cutoffs varying from 6 to 20 mU/L of whole blood.81 Predictably, the lower the cutoff, the greater the number of children labeled as having CH. However, the additional cases identified have mostly functional disorders whose impact on cognition and behavior, if left untreated, is unknown.82
False-negative results are very rare and most are due to errors in specimen handling, testing and data analysis, or reporting of results. Some jurisdictions obtain screening samples in addition to the initial one in low-birth-weight infants, based on observations of a delayed TSH rise in some.83,84 The mechanisms for such delayed rises are unknown, but the widespread use of dopamine infusions, a potent inhibitor of TSH secretion, in the first few days of life may play a role.85 An update on a subset of low-birth-weight newborns with a delayed TSH rise has shown that the problem was transient, with no evidence of benefit from treatment.86 Thus, the repeat screening policy in low-birth-weight newborns remains debatable.87 By contrast, the case for a repeat screening policy in same-sex twins risk is stronger, as fetal blood mixing may lead to a falsely negative screening TSH in the affected member of a monozygotic twin pair.88
As is obvious from the previous discussion, estimating the prevalence of CH depends on the ascertainment method. Before population-based biochemical screening of neonates, it was 1 in 6700 births.89 Over the almost four decades of the screening era, it has increased progressively from 1 in 4000 to 1 in 2500 with our current screening algorithm 82 or even 1 in 1500 in some jurisdictions.90,91 Geographical and temporal variations are due almost exclusively to differences or changes in screening algorithms.92 In keeping with this concept, permanent CH due to thyroid dysgenesis documented by radionuclide imaging is not increasing, whereas the prevalence of dyshormonogenesis, a group of autosomal recessive conditions, is predictably higher in inbred populations93 or in those with a founder effect.94 Interestingly, the introduction of folic acid fortification, which has resulted in a marked decrease in the prevalence of neural tube defects95 and of some heart malformations,96 has not affected the prevalence of CH due to thyroid dysgenesis in the same jurisdiction.82 Universal newborn screening has also provided estimates of the relative birth prevalence of various congenital thyroid disorders at the population level (Table 7-1).
TABLE 7-1
Estimated Birth Prevalence of Various Types of Permanent Thyroid Disorders in Iodine-Sufficient Populations, Based on Universal Newborn Screening with Either TSH or T4
| Type of Permanent Congenital Disorder | Estimated Birth Prevalence |
| Thyroid dysgenesis —Ectopy —Athyreosis | 1:5,000 1:15,000 |
| Thyroid dyshormonogenesis | 1:30,000 |
| Hypothalamic-pituitary hypothyroidism | 1:21,000 |
| Thyroid hormone resistance | 1:40,000 |
From Deladoey J, Ruel J, Giguere Y, Van Vliet G (2011). Is the incidence of congenital hypothyroidism really increasing? A 20-year retrospective population-based study in Quebec. J Clin Endocrinol Metab 96:2422-2429; Kempers MJ, Lanting CI, van Heijst AF, et al. (2006). Neonatal screening for congenital hypothyroidism based on thyroxine, thyrotropin, and thyroxine-binding globulin measurement: potentials and pitfalls. J Clin Endocrinol Metab 91:3370-3376; Lafranchi SH, Snyder DB, Sesser DE, et al. (2003). Follow-up of newborns with elevated screening T4 concentrations. J Pediatr 143:296-301; Tajima T, Jo W, Fujikura K, et al. (2009). Elevated free thyroxine levels detected by a neonatal screening system. Pediatr Res 66: 312-316.
Thyroid dysgenesis
Dysgenesis is defined as abnormal organogenesis, a process that occurs during the embryonic period. The most common developmental defect, currently accounting for about half of CH cases,82 is a defect in the migration of the median anlage, resulting in an ectopic (sub)lingual thyroid. These ectopic thyroids are round structures, lacking lateral lobes, and are the only thyroid tissue present in affected individuals. They can only be unequivocally visualized by radionuclide scanning.97 In almost 10% of cases, there is a dumbbell appearance, with two adjacent areas of ectopic tissue.98 Histologically, ectopic thyroids are fully differentiated with a normal follicular architecture. Thus, the associated hypothyroidism, which is variable in severity, is likely due to a smaller number of cells, due to the absence of lateral lobes, and to a limitation to TSH-induced cell growth.99 The second category of thyroid dysgenesis, athyreosis, is heterogeneous. Indeed, about half of newborns with CH and undetectable technetium uptake on imaging have a detectable serum thyroglobulin.100 We have proposed to call this situation “apparent athyreosis.” Apparent athyreosis can be permanent in patients with a severely hypoplastic and hypofunctional orthotopic thyroid due to mutations that completely inactivate the TSH receptor.50 The term transient athyreosis has been used to describe the absent uptake that can be observed in babies with CH due to the transplacental transfer of TSH receptor-blocking antibodies.101
Thyroid dysgenesis is usually sporadic but familial cases occur more often than by chance alone.102 On the other hand, monozygotic twins are almost always discordant.88 To reconcile these seemingly discrepant findings, a two-hit model combining germ-line susceptibility and early postzygotic mutations has been proposed,22 but its validation requires access to ectopic thyroids, which generally do not need to be removed. The limited data available seem to exclude somatic mutations in TTF-1, TTF-2, and PAX-899 and suggest the implication of the Wnt-beta catenin pathway in thyroid migration.103
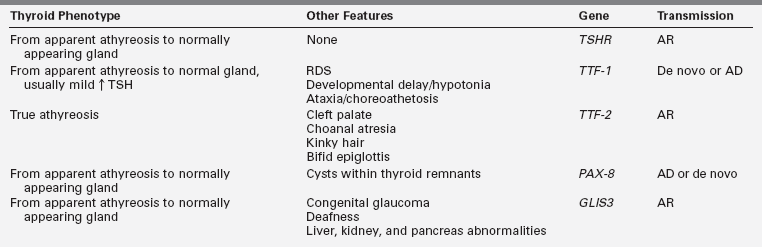
Also inconsistent with simple Mendelian genetics is the observation that thyroid dysgenesis is about twofold more prevalent in female than in male infants. In most series, the skewing of the sex ratio is more pronounced in ectopy than in athyreosis.104 Thyroid dysgenesis is less prevalent in blacks, who are more genetically diverse than Caucasians, and this supports an oligogenic first-hit susceptibility.105 In some of the rare multiplex families, mutations in TTF-1, TTF-2, or PAX8 can be found but these genes have been excluded by linkage analysis in other multiplex families.106 TTF-2 mutations appear to be the least common, having been found in the homozygous state in only three consanguineous pedigrees in which probands present with true athyreosis and cleft palate, sometimes associated with kinky hair, choanal atresia, and a bifid epiglottis.107 TTF-1 heterozygous mutations, de novo or dominantly inherited, present with a spectrum of anomalies, the “brain-lung-thyroid syndrome,” in which CH is usually mild and associated with normal thyroid gland morphology108,109 but may rarely be severe with apparent athyreosis.110 Heterozygous mutations in PAX8 can occur de novo or be inherited in an autosomal dominant fashion.111 They have been associated with hypothyroidism of greatly variable severity and age of onset, even within the same pedigree, with either apparent athyreosis or orthotopic thyroid hypoplasia. In spite of the ubiquitous expression of PAX8, the phenotype is generally limited to hypothyroidism.112 Lastly, mutations of another transcription factor (GL1S3) have been described as the cause of a rare syndrome of CH with apparent athyreosis or normal thyroid anatomy, neonatal diabetes, congenital glaucoma and deafness, liver, kidney, and pancreas abnormalities.113,114 Thus, all transcription factor mutations in humans have been found to cause true athyreosis (TTF2), apparent athyreosis, or orthotopic thyroid hypoplasia (TTF1, PAX8, GLIS3) (Table 7-2) but thyroid ectopy, the most common form of thyroid dysgenesis, remains unexplained.
TABLE 7-2
Thyroid and Extrathyroid Phenotypes Associated with TSHR and Transcription Factor Mutations and Mode of Inheritance
Newborns with trisomy 21, as a group, have a slight skewing of their T4 distribution toward low values and of TSH toward higher values.115 However, these shifts are very subtle so that TSH rarely exceeds screening cutoffs. In fact, not a single case of CH was found in a newborn with trisomy 21 over 20 years in Québec (1.6 million births), so that previous statements that the prevalence of CH is markedly increased in trisomy 21 are clearly unfounded.82
An increased prevalence of extrathyroid abnormalities has consistently been found in several large series; specifically, defects in heart septation are observed in about 5% of dysgenetic CH cohorts.104,116,117 The molecular mechanisms underlying this association are unknown. These septal defects are usually mild and close spontaneously so that, beyond a careful clinical examination, they do not need to be searched for by other tests.
Overt CH due to defects in TSH binding/action resulting in severe thyroid hypoplasia at birth18 represents a developmental disorder (growth being part of development), and we therefore think it is more appropriate to discuss this entity under dysgenesis rather than under dyshormonogenesis. Severe TSH resistance was postulated in 1968118 and the molecular characterization of the TSH receptor, a member of the family of G-protein–coupled receptors,119 has led to the identification of homozygote or compound heterozygote loss-of-function mutations of the TSH receptor gene in several patients with this phenotype.18,50,120 However, the phenotype resulting from biallelic TSH receptor inactivating mutations is generally milder.121,

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


